SARS-CoV-2 and COVID-19: An Evolving Review of Diagnostics and Therapeutics
This manuscript
(permalink)
was automatically generated
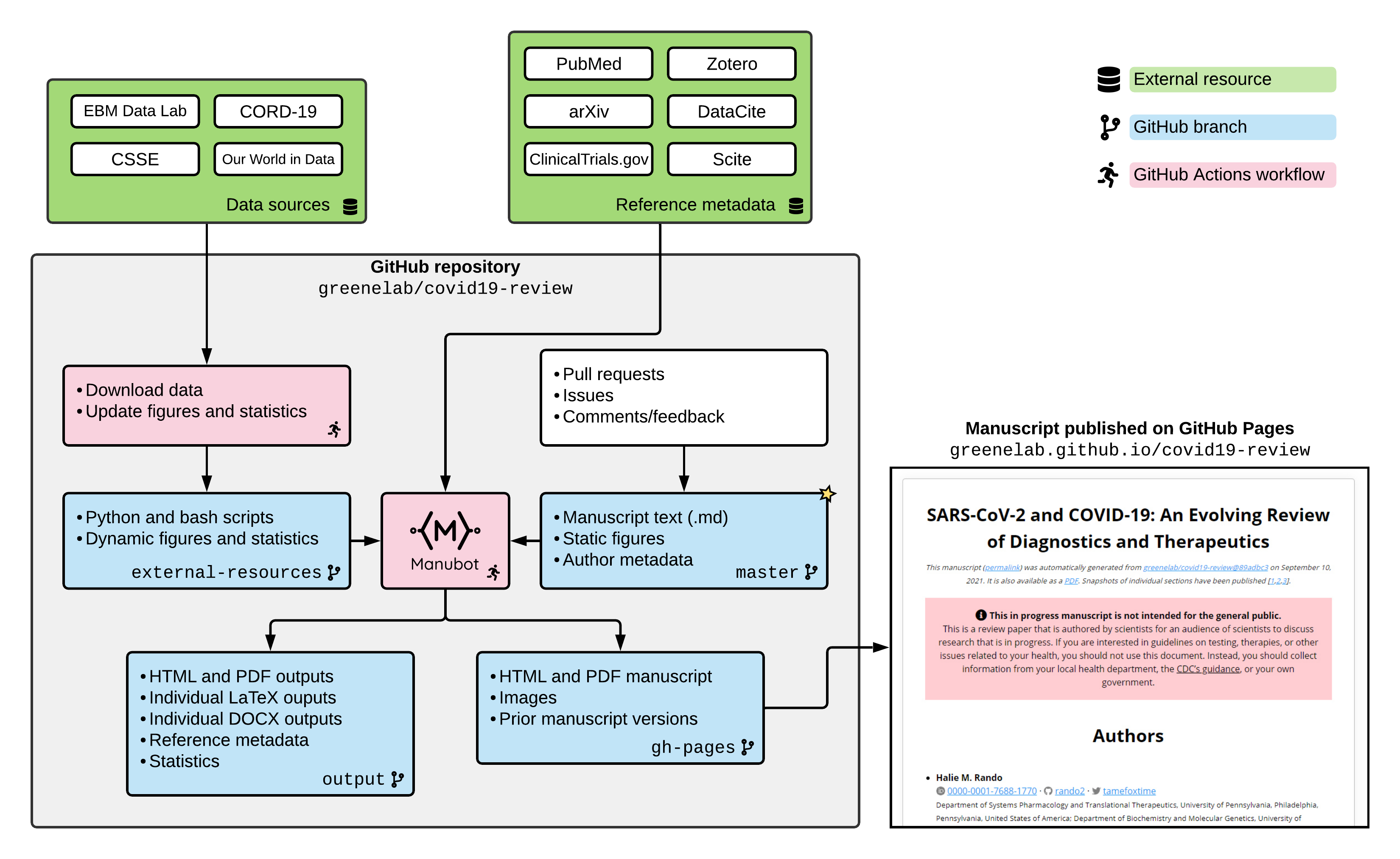
from greenelab/covid19-review@3186561
on February 2, 2024.
It is also available as a PDF.
Snapshots of individual sections have been published (1–6) or posted as preprints (7).
This in progress manuscript is not intended for the general public.
This is a review paper that is authored by scientists for an audience of scientists to discuss research that is in progress.
If you are interested in guidelines on testing, therapies, or other issues related to your health, you should not use this document.
Instead, you should collect information from your local health department, the CDC’s guidance, or your own government.
This project was most active from March 2020 through February 2023 and is not currently receiving major updates.
Authors
Halie M. Rando 0000-0001-7688-1770 · rando2 · tamefoxtime
Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America; Department of Biochemistry and Molecular Genetics, University of Colorado Anschutz School of Medicine, Aurora, Colorado, United States of America; Center for Health AI, University of Colorado Anschutz School of Medicine, Aurora, Colorado, United States of America; Department of Biomedical Informatics, University of Colorado Anschutz School of Medicine, Aurora, Colorado, United States of America
· Funded by the Gordon and Betty Moore Foundation (GBMF 4552); the National Human Genome Research Institute (R01 HG010067)
Casey S. Greene 0000-0001-8713-9213 · cgreene · GreeneScientist
Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America; Childhood Cancer Data Lab, Alex’s Lemonade Stand Foundation, Philadelphia, Pennsylvania, United States of America; Department of Biochemistry and Molecular Genetics, University of Colorado Anschutz School of Medicine, Aurora, Colorado, United States of America; Center for Health AI, University of Colorado Anschutz School of Medicine, Aurora, Colorado, United States of America; Department of Biomedical Informatics, University of Colorado Anschutz School of Medicine, Aurora, Colorado, United States of America
· Funded by the Gordon and Betty Moore Foundation (GBMF 4552); the National Human Genome Research Institute (R01 HG010067)
Michael P. Robson 0000-0002-4859-0033 · mprobson
Department of Computing Sciences, Villanova University, Villanova, Pennsylvania, United States of America
Simina M. Boca 0000-0002-1400-3398 · SiminaB
Innovation Center for Biomedical Informatics, Georgetown University Medical Center, Washington, District of Columbia, United States of America
Nils Wellhausen 0000-0001-8955-7582 · nilswellhausen
Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America
Ronan Lordan 0000-0001-9668-3368 · RLordan · el_ronan
Institute for Translational Medicine and Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104-5158, USA; Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA; Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania; Philadelphia, PA 19104, USA
Christian Brueffer 0000-0002-3826-0989 · cbrueffer · cbrueffer
InSilico Consulting AB, Lund, Sweden; Department of Clinical Sciences, Lund University, Lund, Sweden
Sandipan Ray 0000-0002-9960-5768 · rays1987
Department of Biotechnology, Indian Institute of Technology Hyderabad, Kandi, Sangareddy 502285, Telangana, India
Lucy D'Agostino McGowan 0000-0001-7297-9359 · LucyMcGowan · LucyStats
Department of Mathematics and Statistics, Wake Forest University, Winston-Salem, North Carolina, United States of America
Anthony Gitter 0000-0002-5324-9833 · agitter · anthonygitter
Department of Biostatistics and Medical Informatics, University of Wisconsin-Madison, Madison, Wisconsin, United States of America; Morgridge Institute for Research, Madison, Wisconsin, United States of America
· Funded by John W. and Jeanne M. Rowe Center for Research in Virology
Anna Ada Dattoli 0000-0003-1462-831X · aadattoli · aadattoli
Department of Pathology and Laboratory Medicine, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Department of Systems Pharmacology & Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
John P. Barton 0000-0003-1467-421X · johnbarton · _jpbarton
Department of Physics and Astronomy, University of California-Riverside, Riverside, California, United States of America
Jeffrey M. Field 0000-0001-7161-7284 · Jeff-Field
Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
Adam L. MacLean 0000-0003-0689-7907 · adamlmaclean · adamlmaclean
Department of Quantitative and Computational Biology, University of Southern California, Los Angeles, California, United States of America
Alexandra J. Lee 0000-0002-0208-3730 · ajlee21
Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America
· Funded by the Gordon and Betty Moore Foundation (GBMF 4552)
Immunology Institute of the Icahn School of Medicine · ismms-himc
Immunology Institute of the Icahn School of Medicine
Fengling Hu 0000-0003-1081-5038 · hufengling · hufengling
Department of Biostatistics, Epidemiology and Informatics, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America
Nafisa M. Jadavji 0000-0002-3557-7307 · nafisajadavji · nafisajadavji
Biomedical Science, Midwestern University, Glendale, AZ, United States of America; Department of Neuroscience, Carleton University, Ottawa, Ontario, Canada
· Funded by the American Heart Association (20AIREA35050015)
Elizabeth Sell 0000-0002-9658-1107 · esell17
Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America
Vincent Rubinetti 0000-0002-4655-3773 · vincerubinetti
Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America; Center for Health AI, University of Colorado School of Medicine, Aurora, Colorado, United States of America
Jinhui Wang 0000-0002-5796-8130 · jinhui2
Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America
Diane N. Rafizadeh 0000-0002-2838-067X · dianerafi
Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America; Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America
· Funded by NIH Medical Scientist Training Program T32 GM07170
Ashwin N. Skelly 0000-0002-1565-3376 · anskelly
Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America; Institute for Immunology, University of Pennsylvania Perelman School of Medicine, Philadelphia, United States of America
· Funded by NIH Medical Scientist Training Program T32 GM07170
Marouen Ben Guebila 0000-0001-5934-966X · marouenbg · marouenbg
Department of Biostatistics, Harvard School of Public Health, Boston, Massachusetts, United States of America
Likhitha Kolla 0000-0002-1169-906X · likhithakolla · lkolla2018
Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America
· Funded by NIH Medical Scientist Training Program T32 GM07170
David Manheim 0000-0001-8599-8380 · davidmanheim · davidmanheim
1DaySooner, Delaware, United States of America; Risk and Health Communication Research Center, School of Public Health, University of Haifa, Haifa, Israel; Technion, Israel Institute of Technology, Haifa, Israel
· Funded by Center for Effective Altruism, Long Term Future Fund
Soumita Ghosh 0000-0002-2783-2750 · soumitagh
Institute of Translational Medicine and Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America
James Brian Byrd 0000-0002-0509-3520 · byrdjb · thebyrdlab
University of Michigan School of Medicine, Ann Arbor, Michigan, United States of America
· Funded by NIH K23HL128909; FastGrants
YoSon Park 0000-0002-0465-4744 · ypar · yoson
Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America
· Funded by NHGRI R01 HG10067
Vikas Bansal 0000-0002-0944-7226 · bansalvi · VikasBansal1989
Biomedical Data Science and Machine Learning Group, German Center for Neurodegenerative Diseases, Tübingen 72076, Germany
Stephen Capone 0000-0001-7231-1535 · scapone01
St. George’s University School of Medicine, St. George’s, Grenada
John J. Dziak 0000-0003-0762-5495 · dziakj1
Edna Bennett Pierce Prevention Research Center, The Pennsylvania State University, University Park, PA, United States of America
Yuchen Sun · kevinsunofficial
Department of Computer Science, University of Virginia, Charlottesville, VA, United States of America
Yanjun Qi 0000-0002-5796-7453 · qiyanjun
Department of Computer Science, University of Virginia, Charlottesville, VA, United States of America
Lamonica Shinholster 0000-0001-6285-005X · LSH2126
Mercer University, Macon, GA, United States of America
· Funded by the Center for Global Genomics and Health Equity at the University of Pennsylvania
Temitayo Lukan · tlukan
University of Pennsylvania, Philadelphia, PA, United States of America
Dimitri Perrin 0000-0002-4007-5256 · SystemsResearch · dperrin
School of Computer Science, Queensland University of Technology, Brisbane, Australia; Centre for Data Science, Queensland University of Technology, Brisbane, Australia
Serghei Mangul 0000-0003-4770-3443 · smangul1 · serghei_mangul
Department of Clinical Pharmacy, School of Pharmacy, University of Southern California, Los Angeles, CA, United States of America
Shikta Das 0000-0002-8291-2788 · shiktadas · shikta_das
C4X Discovery, London, United Kingdom; Medical Research Council LHA, Institute of Cardiovascular Studies, University College London, London, United Kingdom
Tiago Lubiana 0000-0003-2473-2313 · lubianat · lubianat
Department of Clinical and Toxicological Analyses, School of Pharmaceutical Sciences, University of São Paulo, São Paulo, Brazil
David Mai 0000-0002-9238-0164 · davemai · lococyte
Department of Bioengineering, University of Pennsylvania, Philadelphia, PA, USA; Center for Cellular Immunotherapies, Perelman School of Medicine, and Parker Institute for Cancer Immunotherapy at University of Pennsylvania, Philadelphia, PA, USA
Joel D Boerckel 0000-0003-3126-3025 · jboerckel · jboerckel
Department of Orthopaedic Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States of America; Department of Bioengineering, University of Pennsylvania, Philadelphia, PA, United States of America
Amruta Naik 0000-0003-0673-2643 · NAIKA86
Children’s Hospital of Philadelphia, Philadelphia, PA, United States of America
Yusha Sun 0000-0003-4835-3000 · yusha-sun
Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America
Daniel S. Himmelstein 0000-0002-3012-7446 · dhimmel · dhimmel
Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America; Related Sciences
· Funded by GBMF4552
Jeremy P. Kamil 0000-0001-8422-7656
Department of Microbiology and Immunology, Louisiana State University Health Sciences Center Shreveport, Shreveport, Louisiana, USA
Jesse G. Meyer 0000-0003-2753-3926 · jessegmeyerlab
Department of Biochemistry, Medical College of Wisconsin, Milwaukee, Wisconsin, United States of America
· Funded by National Institute of General Medical Sciences (R35 GM142502)
Ariel I. Mundo 0000-0002-6014-4538 · aimundo
Department of Biomedical Engineering, University of Arkansas, Fayetteville, Arkansas, USA
COVID-19 Review Consortium:
Vikas Bansal, John P. Barton, Simina M. Boca, Joel D Boerckel, Christian Brueffer, James Brian Byrd, Stephen Capone, Shikta Das, Anna Ada Dattoli, John J. Dziak, Jeffrey M. Field, Soumita Ghosh, Anthony Gitter, Rishi Raj Goel, Casey S. Greene, Marouen Ben Guebila, Daniel S. Himmelstein, Fengling Hu, Nafisa M. Jadavji, Jeremy P. Kamil, Sergey Knyazev, Likhitha Kolla, Alexandra J. Lee, Ronan Lordan, Tiago Lubiana, Temitayo Lukan, Adam L. MacLean, David Mai, Serghei Mangul, David Manheim, Lucy D'Agostino McGowan, Jesse G. Meyer, Ariel I. Mundo, Amruta Naik, YoSon Park, Dimitri Perrin, Yanjun Qi, Diane N. Rafizadeh, Bharath Ramsundar, Halie M. Rando, Sandipan Ray, Michael P. Robson, Vincent Rubinetti, Elizabeth Sell, Lamonica Shinholster, Ashwin N. Skelly, Yuchen Sun, Yusha Sun, Gregory L Szeto, Ryan Velazquez, Jinhui Wang, Nils Wellhausen
Authors are ordered arbitrarily.
1 Pathogenesis, Symptomatology, and Transmission of SARS-CoV-2 through Analysis of Viral Genomics and Structure
1.1 Abstract
The novel coronavirus SARS-CoV-2, which emerged in late 2019, has since spread around the world and infected hundreds of millions of people with coronavirus disease 2019 (COVID-19).
While this viral species was unknown prior to January 2020, its similarity to other coronaviruses that infect humans has allowed for rapid insight into the mechanisms that it uses to infect human hosts, as well as the ways in which the human immune system can respond.
Here, we contextualize SARS-CoV-2 among other coronaviruses and identify what is known and what can be inferred about its behavior once inside a human host.
Because the genomic content of coronaviruses, which specifies the virus’s structure, is highly conserved, early genomic analysis provided a significant head start in predicting viral pathogenesis and in understanding potential differences among variants.
The pathogenesis of the virus offers insights into symptomatology, transmission, and individual susceptibility.
Additionally, prior research into interactions between the human immune system and coronaviruses has identified how these viruses can evade the immune system’s protective mechanisms.
We also explore systems-level research into the regulatory and proteomic effects of SARS-CoV-2 infection and the immune response.
Understanding the structure and behavior of the virus serves to contextualize the many facets of the COVID-19 pandemic and can influence efforts to control the virus and treat the disease.
1.2 Importance
COVID-19 involves a number of organ systems and can present with a wide range of symptoms.
From how the virus infects cells to how it spreads between people, the available research suggests that these patterns are very similar to those seen in the closely related viruses SARS-CoV-1 and possibly MERS-CoV.
Understanding the pathogenesis of the SARS-CoV-2 virus also contextualizes how the different biological systems affected by COVID-19 connect.
Exploring the structure, phylogeny, and pathogenesis of the virus therefore helps to guide interpretation of the broader impacts of the virus on the human body and on human populations.
For this reason, an in-depth exploration of viral mechanisms is critical to a robust understanding of SARS-CoV-2 and, potentially, future emergent HCoV.
1.3 Introduction
The current coronavirus disease 2019 (COVID-19) pandemic, caused by the Severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) virus, represents an acute global health crisis.
Symptoms of the disease can range from mild to severe or fatal (8) and can affect a variety of organs and systems (9).
Outcomes of infection can include acute respiratory distress (ARDS) and acute lung injury, as well as damage to other organ systems (9, 10).
Understanding the progression of the disease, including these diverse symptoms, depends on understanding how the virus interacts with the host.
Additionally, the fundamental biology of the virus can provide insights into how it is transmitted among people, which can, in turn, inform efforts to control its spread.
As a result, a thorough understanding of the pathogenesis of SARS-CoV-2 is a critical foundation on which to build an understanding of COVID-19 and the pandemic as a whole.
The rapid identification and release of the genomic sequence of the virus in January 2020 (11) provided early insight into the virus in a comparative genomic context.
The viral genomic sequence clusters with known coronaviruses (order Nidovirales, family Coronaviridae, subfamily Orthocoronavirinae).
Phylogenetic analysis of the coronaviruses reveals four major subclades, each corresponding to a genus: the alpha, beta, gamma, and delta coronaviruses.
Among them, alpha and beta coronaviruses infect mammalian species, gamma coronaviruses infect avian species, and delta coronaviruses infect both mammalian and avian species (12).
The novel virus now known as SARS-CoV-2 was identified as a beta coronavirus belonging to the B lineage based on phylogenetic analysis of a polymerase chain reaction (PCR) amplicon fragment from five patients along with the full genomic sequence (13).
This lineage also includes the Severe acute respiratory syndrome-related coronavirus (SARS-CoV-1) that caused the 2002-2003 outbreak of Severe Acute Respiratory Syndrome (SARS) in humans (13).
(Note that these subclades are not to be confused with variants of concern within SARS-CoV-2 labeled with Greek letters; i.e., the Delta variant of SARS-CoV-2 is still a beta coronavirus.)
Because viral structure and mechanisms of pathogenicity are highly conserved within the order, this phylogenetic analysis provided a basis for forming hypotheses about how the virus interacts with hosts, including which tissues, organs, and systems would be most susceptible to SARS-CoV-2 infection.
Coronaviruses that infect humans (HCoV) are not common, but prior research into other HCoV such as SARS-CoV-1 and Middle East respiratory syndrome-related coronavirus (MERS-CoV), as well as other viruses infecting humans such as a variety of influenza species, established a strong foundation that accelerated the pace of SARS-CoV-2 research.
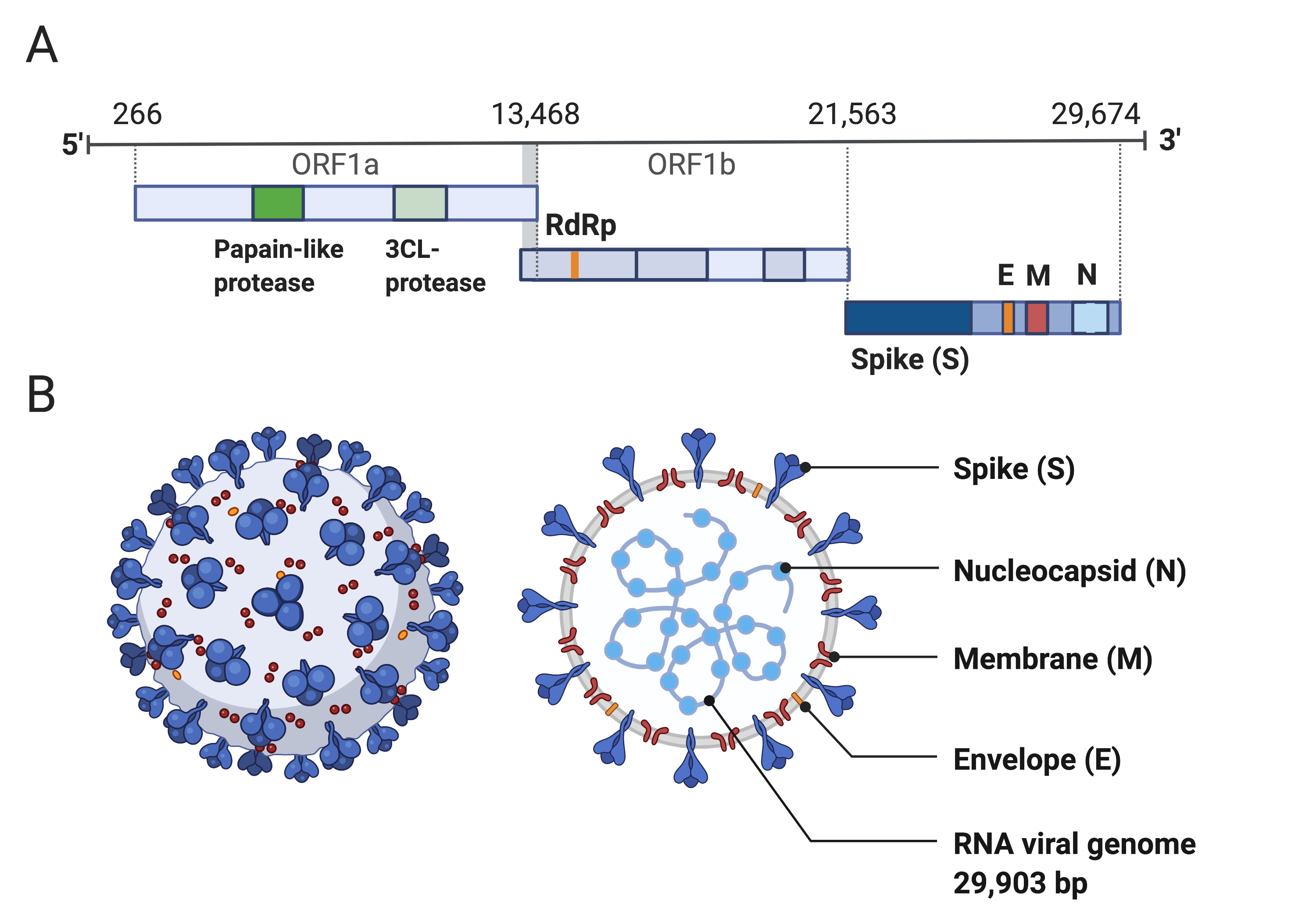
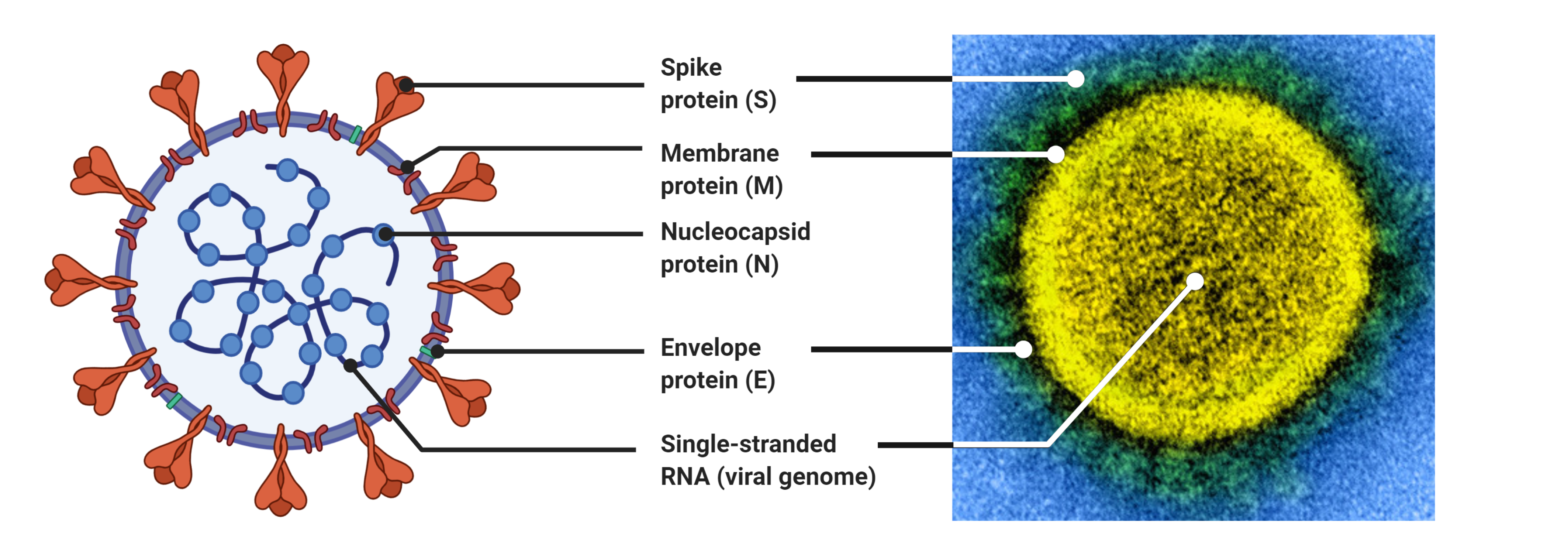
Coronaviruses are large viruses that can be identified by their distinctive “crown-like” shape (Figure 1).
Their spherical virions are made from lipid envelopes ranging from 100 to 160 nanometers in which peplomers (protruding structures) of two to three spike (S) glycoproteins are anchored, creating the crown (14, 15).
These spikes, which are critical to both viral pathogenesis and to the response by the host immune response, have been visualized using cryo-electron microscopy (16).
Because they induce the human immune response, they are also the target of many proposed therapeutic agents (2, 3).
Viral pathogenesis is typically broken down into three major components: entry, replication, and spread (17).
However, in order to draw a more complete picture of pathogenesis, it is also necessary to examine how infection manifests clinically, identify systems-level interactions between the virus and the human body, and consider the possible effects of variation or evolutionary change on pathogenesis and virulence.
Thus, clinical medicine and traditional biology are both important pieces of the puzzle of SARS-CoV-2 presentation and pathogenesis.
1.4 Coronavirus Structure and Pathogenesis
1.4.1 Structure of Coronaviruses
Genome structure is highly conserved among coronaviruses, meaning that the relationship between the SARS-CoV-2 genome and its pathogenesis can be inferred from prior research in related viral species.
The genomes of viruses in the Nidovirales order share several fundamental characteristics.
They are non-segmented, which means the viral genome is a single continuous strand of RNA, and are enveloped, which means that the genome and capsid are encased by a lipid bilayer.
Coronaviruses have large positive-sense RNA (ssRNA+) genomes ranging from 27 to 32 kilobases in length (18, 19).
The SARS-CoV-2 genome lies in the middle of this range at 29,903 bp (19).
Genome organization is highly conserved within the order (18).
There are three major genomic regions: one containing the replicase gene, one containing the genes encoding structural proteins, and interspersed accessory genes (18) (Figure 1).
The replicase gene comprises about two-thirds of the genome and consists of two open reading frames that are translated with ribosomal frameshifting (18).
This polypeptide is then translated into 16 non-structural proteins (nsp), except in gammacoronaviruses where nsp1 is absent, that form the replication machinery used to synthesize viral RNA (20).
The remaining third of the genome encodes structural proteins, including the spike (S), membrane, envelope, and nucleocapsid proteins.
Additional accessory genes are sometimes present between these two regions, depending on the species or strain.
Much attention has been focused on the S protein, which is a critical structure involved in cell entry.
Figure 1:Structure of SARS-CoV-2 capsid and genome.
A) The genomic structure of coronaviruses is highly conserved and includes three main regions.
Open reading frames (ORF) 1a and 1b contain two polyproteins that encode the non-structural proteins (nsp).
The nsp include enzymes such as RNA-dependent RNA Polymerase (RdRp).
The last third of the genome encodes structural proteins, including the spike (S), envelope (E), membrane (M) and nucleocapsid (N) proteins.
Accessory genes can also be interspersed throughout the genome (18).
B) The physical structure of the coronavirus virion, including the components determined by the conserved structural proteins S, E, M and N.
This figure was adapted from “Human Coronavirus Structure”, by BioRender.com (2020), retrieved from https://app.biorender.com/biorender-templates.
1.4.2 Pathogenic Mechanisms of Coronaviruses
While it is possible that SARS-CoV-1 and SARS-CoV-2, like most viruses, enter cells through endocytosis, a process conserved among coronaviruses enables them to target cells for entry through fusion with the plasma membrane (21, 22).
Cell entry proceeds in three steps: binding, cleavage, and fusion.
First, the viral spike protein binds to a host cell via a recognized receptor or entry point.
Coronaviruses can bind to a range of host receptors (23, 24), with binding conserved only at the genus level (12).
Viruses in the beta coronavirus genus, to which SARS-CoV-2 belongs, are known to bind to the CEACAM1 protein, 5-N-acetyl-9-O-acetyl neuraminic acid, and to angiotensin-converting enzyme 2 (ACE2) (23).
This recognition is driven by domains in the S1 subunit (25).
SARS-CoV-2 has a high affinity for human ACE2, which is expressed in the vascular epithelium, other epithelial cells, and cardiovascular and renal tissues (26, 27), as well as many others (28).
The binding process is guided by the molecular structure of the spike protein, which is structured in three segments: an ectodomain, a transmembrane anchor, and an intracellular tail (29).
The ectodomain forms the crown-like structures on the viral membrane and contains two subdomains known as the S1 and S2 subunits (30).
The S1 (N-terminal) domain forms the head of the crown and contains the receptor binding motif, and the S2 (C-terminal) domain forms the stalk that supports the head (30).
The S1 subunit guides the binding of the virus to the host cell, and the S2 subunit guides the fusion process (29).
After the binding of the S1 subunit to an entry point, the spike protein of coronaviruses is often cleaved at the S1/S2 boundary into the S1 and S2 subunits by a host protease (25, 31, 32).
This proteolytic priming is important because it prepares the S protein for fusion (31, 32).
The two subunits remain bound by van der Waals forces, with the S1 subunit stabilizing the S2 subunit throughout the membrane fusion process (25).
Cleavage at a second site within S2 (S2’) activates S for fusion by inducing conformational changes (25).
Similar to SARS-CoV-1, SARS-CoV-2 exhibits redundancy in which host proteases can cleave the S protein (33).
Both transmembrane protease serine protease-2 (TMPRSS-2) and cathepsins B/L have been shown to mediate SARS-CoV-2 S protein proteolytic priming, and small molecule inhibition of these enzymes fully inhibited viral entry in vitro(33, 34).
Other proteases known to cleave the S1/S2 boundary in coronaviruses include TMPRSS-4, trypsin, furin, cathepsins, and human airway trypsin-like protease (HAT) (34).
Unlike in SARS-CoV-1, a second cleavage site featuring a furin-like binding motif is also present near the S1/S2 boundary in SARS-CoV-2 (35).
This site is found in HCoV belonging to the A and C lineages of beta coronavirus, including MERS-CoV, but not in the other known members of the B lineage of beta coronavirus that contains SARS-CoV-1 and SARS-CoV-2 (35).
It is associated with increased virulence in other viral species (35) and may facilitate membrane fusion of SARS-CoV-2 in the absence of other proteases that prime the S1/S2 site (36).
However, given that proteases such as HAT are likely to be present in targets like the human airway, the extent to which this site has had a real-world effect on the spread of SARS-CoV-2 was initially unclear (36).
Subsequent research has supported this site as an important contributor to pathogenesis: in vitro analyses have reported that it bolsters pathogenicity specifically in cell lines derived from human airway cells (Calu3 cell line) (37–39) and that furin inhibitors reduced pathogenic effects in VeroE6 cells (40).
Electron microscopy suggests that in some coronaviruses, including SARS-CoV-1 and MERS-CoV, a six-helix bundle separates the two subunits in the postfusion conformation, and the unusual length of this bundle facilitates membrane fusion through the release of additional energy (12).
The viral membrane can then fuse with the endosomal membrane to release the viral genome into the host cytoplasm.
Once the virus enters a host cell, the replicase gene is translated and assembled into the viral replicase complex.
This complex then synthesizes the double-stranded RNA (dsRNA) genome from the genomic ssRNA(+).
The dsRNA genome is transcribed and replicated to create viral mRNAs and new ssRNA(+) genomes (18, 41).
From there, the virus can spread into other cells.
In SARS-CoV-2, the insertion of the furin-like binding site near the S1/S2 boundary is also thought to increase cell-cell adhesion, making it possible for the viral genome to spread directly from cell to cell rather than needing to propagate the virion itself (42).
In this way, the genome of SARS-CoV-2 provides insight into the pathogenic behavior of the virus.
Evidence also suggests that SARS-CoV-2 may take advantage of the specific structure of endothelial cells to enter the circulatory system.
Endothelial cells are specialized epithelial cells (43) that form a barrier between the bloodstream and surrounding tissues.
The endothelium facilitates nutrient, oxygen, and cellular exchange between the blood and vascularized tissues (44).
The luminal (interior) surface of the endothelium is lined with glycocalyx, a network of both membrane-bound and soluble proteins and carbohydrates, primarily proteoglycans and glycoproteins (45, 46).
The glycocalyx varies in thickness from 0.5 microns in the capillaries to 4.5 microns in the carotid arteries and forms a meshwork that localizes both endothelial- and plasma-derived signals to the inner vessel wall (45).
Heparan sulfate is the dominant proteoglycan in the glycocalyx, representing 50-90% of glycocalyx proteoglycan content (47).
The SARS-CoV-2 spike protein can bind directly to heparan sulfate, which serves in part as a scaffolding molecule to facilitate ACE2 binding and entry into endothelial cells (46).
A heparan sulfate binding site has also been identified near the ACE2 binding site on the viral receptor binding domain (RBD), and modeling has suggested that heparan sulfate binding yields an open conformation that facilitates binding to ACE2 on the cell surface (46).
Degrading or removing heparan sulfate was associated with decreased binding (46).
Heparan sulfate may also interact with the S1/S2 proteolytic cleavage site and other binding sites to promote binding affinity (48).
Notably, treatment with soluble heparan sulfate or even heparin (a commonly used anti-coagulant and vasodilator that is similar in structure to heparan sulfate (49)) potently blocked spike protein binding and viral infection (46).
This finding is particularly interesting because degradation of heparan sulfate in the glycocalyx has previously been identified as an important contributor to ARDS and sepsis (50), two common and severe outcomes of COVID-19, and suggests that heparan sulfate could be a target for pharmaceutical inhibition of cell entry by SARS-CoV-2 (51–55).
Together, this evidence suggests that heparan sulfate can serve as an important adhesion molecule for SARS-CoV-2 cell entry.
It may represent a therapeutic target but has not been pursued as much as other candidate targets (3).
1.4.3 Immune Evasion Strategies
Research in other HCoV provides some indication of how SARS-CoV-2 infection can proceed despite human immune defenses.
Infecting the epithelium can help viruses such as SARS-CoV-1 bypass the physical barriers, such as mucus, that comprise the immune system’s first line of defense (56).
Once the virus infiltrates host cells, it is adept at evading detection.
CD163+ and CD68+ macrophage cells are especially crucial for the establishment of SARS-CoV-1 in the body (56).
These cells most likely serve as viral reservoirs that help shield SARS-CoV-1 from the innate immune response.
According to a study on the viral dissemination of SARS-CoV-1 in Chinese macaques, viral RNA could be detected in some monocytes throughout the process of differentiation into dendritic cells (56).
This lack of active viral replication allows SARS-CoV-1 to escape the innate immune response because reduced levels of detectable viral RNA allow the virus to avoid both natural killer cells and Toll-like receptors (56).
Even during replication, SARS-CoV-1 is able to mask its dsRNA genome from detection by the immune system.
Although dsRNA is a pathogen-associated molecular pattern that would typically initiate a response from the innate immune system (57), in vitro analysis of nidoviruses including SARS-CoV-1 suggests that these viruses can induce the development of double-membrane vesicles that protect the dsRNA signature from being detected by the host immune system (58).
This protective envelope can therefore insulate these coronaviruses from the innate immune system’s detection mechanism (59).
HCoVs are also known to interfere with the host immune response, rather than just evade it.
For example, the virulence of SARS-CoV-2 is increased by nsp1, which can suppress host gene expression by stalling mRNA translation and inducing endonucleolytic cleavage and mRNA degradation (60).
SARS-CoV-1 also evades the immune response by interfering with type I IFN induction signaling, which is a mechanism that leads to cellular resistance to viral infections.
SARS-CoV-1 employs methods such as ubiquitination and degradation of RNA sensor adaptor molecules MAVS and TRAF3/6 (61).
Also, MERS-CoV downregulates antigen presentation via MHC class I and MHC class II, which leads to a reduction in T cell activation (61).
These evasion mechanisms, in turn, may facilitate systemic infection.
Coronaviruses such as SARS-CoV-1 are also able to evade the humoral immune response through other mechanisms, such as inhibiting certain cytokine pathways or down-regulating antigen presentation by the cells (58).
1.4.4 Host Cell Susceptibility
ACE2 and TMPRSS-2 have been identified as the primary entry portal and as a critical protease, respectively, in facilitating the entry of SARS-CoV-1 and SARS-CoV-2 into a target cell (16, 33, 62–64).
This finding has led to a hypothesized role for the expression of these molecules in determining which cells, tissues, and organs are most susceptible to SARS-CoV-2 infection.
ACE2 is expressed in numerous organs, such as the heart, kidney, and intestine, but it is most prominently expressed in alveolar epithelial cells; this pattern of expression is expected to contribute to the virus’ association with lung pathology (26, 65, 66) as well as that of SARS (67).
A retrospective observational study reported indirect evidence that certain antineoplastic therapies, such as the chemotherapy drug gemcitabine, may reduce risk of SARS-CoV-2 infection in patients with cancer, possibly via decreased ACE2 expression (68).
Additionally, the addition of the furin site insertion at the S1/S2 boundary means that SARS-CoV-2 does not require TMPRSS-2 when furin, an ubiquitously expressed endoprotease (69), is present, enabling cell-cell fusion independent of TMPRSS-2 availability (70).
Clinical investigations of COVID-19 patients have detected SARS-CoV-2 transcripts in bronchoalveolar lavage fluid (BALF) (93% of specimens), sputum (72%), nasal swabs (63%), fibrobronchoscopy brush biopsies (46%), pharyngeal swabs (32%), feces (29%), and blood (1%) (71).
Two studies reported that SARS-CoV-2 could not be detected in urine specimens (71, 72); however, a third study identified four urine samples (out of 58) that were positive for SARS-CoV-2 nucleic acids (73).
Although respiratory failure remains the leading cause of death for COVID-19 patients (74), SARS-CoV-2 infection can damage many other organ systems including the heart (75), kidneys (76, 77), liver (78), and gastrointestinal tract (79, 80).
As it becomes clear that SARS-CoV-2 infection can damage multiple organs, the scientific community is pursuing multiple avenues of investigation in order to build a consensus about how the virus affects the human body.
1.5 Clinical Presentation of COVID-19
SARS-CoV-2 pathogenesis is closely linked with the clinical presentation of the COVID-19 disease.
Reports have described diverse symptom profiles associated with COVID-19, with a great deal of variability both within and between institutions and regions.
Definitions for non-severe, severe, and critical COVID-19, along with treatment recommendations, are available from the World Health Organization living guidelines (81).
A large study from Wuhan, China conducted early in the pandemic identified fever and cough as the two most common symptoms that patients reported at hospital admission (82), while a retrospective study in China described the clinical presentations of patients infected with SARS-CoV-2 as including lower respiratory tract infection with fever, dry cough, and dyspnea (shortness of breath) (83).
This study (83) noted that upper respiratory tract symptoms were less common, suggesting that the virus preferentially targets cells located in the lower respiratory tract.
However, data from the New York City region (84, 85) showed variable rates of fever as a presenting symptom, suggesting that symptoms may not be consistent across individuals.
For example, even within New York City, one study (84) identified low oxygen saturation (<90% without the use of supplemental oxygen or ventilation support) in 20.4% of patients upon presentation, with fever being present in 30.7%, while another study (85) reported cough (79.4%), fever (77.1%), and dyspnea (56.5%) as the most common presenting symptoms; both of these studies considered only hospitalized patients.
A later study reported radiographic findings such as ground-glass opacity and bilateral patchy shadowing in the lungs of many hospitalized patients, with most COVID-19 patients having lymphocytopenia, or low levels of lymphocytes (a type of white blood cell) (82).
Patients may also experience loss of smell, myalgias (muscle aches), fatigue, or headache.
Gastrointestinal symptoms can also present (86), and the CDC includes nausea and vomiting, as well as congestion and runny nose, on its list of symptoms consistent with COVID-19 (8).
An analysis of an app-based survey of 500,000 individuals in the U.S. found that among those tested for SARS-CoV-2, a loss of taste or smell, fever, and a cough were significant predictors of a positive test result (87).
It is important to note that in this study, the predictive value of symptoms may be underestimated if they are not specific to COVID-19.
This underestimation could occur because the outcome measured was a positive, as opposed to a negative, COVID-19 test result, meaning an association would be more easily identified for symptoms that were primarily or exclusively found with COVID-19.
At the time the surveys were conducted, due to limits in U.S. testing infrastructure, respondents typically needed to have some symptoms known to be specific to COVID-19 in order to qualify for testing.
Widespread testing of asymptomatic individuals may therefore provide additional insight into the range of symptoms associated with COVID-19.
Consistent with the wide range of symptoms observed and the pathogenic mechanisms described above, COVID-19 can affect a variety of systems within the body in addition to causing respiratory problems (88).
For example, COVID-19 can lead to acute kidney injury, especially in patients with severe respiratory symptoms or certain preexisting conditions (89).
Some patients are at risk for collapsing glomerulopathy (90).
COVID-19 can also cause neurological complications (91–93), potentially including stroke, seizures or meningitis (94, 95).
One study on autopsy samples suggested that SARS-CoV-2 may be able to enter the central nervous system via the neural–mucosal interface (96).
However, a study of 41 autopsied brains (97) found no evidence that the virus can actually infect the central nervous system.
Although there was viral RNA in some brain samples, it was only found in very small amounts, and no viral protein was found.
The RNA may have been in the blood vessels or blood components and not in the brain tissue itself.
Instead, the neuropathological effects of COVID-19 are more likely to be caused indirectly by hypoxia, coagulopathy, or inflammatory processes rather than by infection in the brain (97).
COVID-19 has been associated with an increased incidence of large vessel stroke, particularly in patients under the age of 40 (98), and other thrombotic events including pulmonary embolism and deep vein thrombosis (99).
The mechanism behind these complications has been suggested to be related to coagulopathy, with reports indicating the presence of antiphospholipid antibodies (100) and elevated levels of d-dimer and fibrinogen degradation products in deceased patients (101).
Other viral infections have been associated with coagulation defects and changes to the coagulation cascade; notably, SARS was also found to lead to disseminated intravascular coagulation and was associated with both pulmonary embolism and deep vein thrombosis (102).
The mechanism behind these insults has been suggested to be related to inflammation-induced increases in the von Willebrand factor clotting protein, leading to a pro-coagulative state (102).
Abnormal clotting (thromboinflammation or coagulopathy) has been increasingly discussed recently as a possible key mechanism in many cases of severe COVID-19, and may be associated with the high d-dimer levels often observed in severe cases (103–105).
This excessive clotting in lung capillaries has been suggested to be related to a dysregulated activation of the complement system, part of the innate immune system (106, 107).
Finally, concerns have been raised about long-term sequelae of COVID-19.
Some COVID-19 patients have reported that various somatic symptoms (such as shortness of breath, fatigue, chest pain) and psychological (depression, anxiety or mild cognitive impairment) symptoms can last for months after infection (108).
Such long-term affects occur both in adults (109) and children (110).
Sustained symptoms affecting a variety of biological systems have been reported across many studies (e.g., (108, 111, 112)).
The phenomenon of “long COVID” is not fully understood although various possible explanations have been proposed, including damage caused by immune response to infection as well as by the infection itself, in addition to negative consequences of the experience of lengthy illness and hospitalization.
However, a lack of consistency among definitions used in different studies makes it difficult to develop precise definitions or identify specific symptoms associated with long-term effects of COVID-19 (113, 114).
Patient and family support groups for “long haulers” have been formed online, and patient-driven efforts to collect data about post-acute COVID-19 provide valuable sources of information (e.g., (111)).
The specific relationship between viral pathogenesis and these reported sequelae remains to be uncovered, however.
1.5.1 Pediatric Presentation
The presentation of COVID-19 infection can vary greatly among pediatric patients and, in some cases, manifests in distinct ways from COVID-19 in adults.
Evidence suggests that children and adolescents tend to have mostly asymptomatic infections and that those who are symptomatic typically exhibit mild illness (115–118).
One review examined symptoms reported in 17 studies of children infected with COVID-19 during the early months of the COVID-19 epidemic in China and one study from Singapore (119).
In the more than a thousand cases described, the most common reports were for mild symptoms such as fever, dry cough, fatigue, nasal congestion and/or runny nose, while three children were reported to be asymptomatic.
Severe lower respiratory infection was described in only one of the pediatric cases reviewed.
Gastrointestinal symptoms such as vomiting or diarrhea were occasionally reported.
Radiologic findings were not always reported in the case studies reviewed, but when they were mentioned they included bronchial thickening, ground-glass opacities, and/or inflammatory lesions (119).
Neurological symptoms have also been reported (120).
These analyses indicate that most pediatric cases of COVID-19 are not severe.
Indeed, it is estimated that less than 1% of pediatric cases result in critical illness (117, 121), although reporting suggests that pediatric hospitalizations may be greater with the emergence of the Delta variant of concern (VOC) (122–124).
Serious complications and, in relatively rare cases, deaths have occurred (125).
Of particular interest, children have occasionally experienced a serious inflammatory syndrome, multisystem inflammatory syndrome in children (MIS-C), following COVID-19 infection (126).
This syndrome is similar in some respects to Kawasaki disease, including Kawasaki disease shock syndrome (127–129), and is thought to be a distinct clinical manifestation of SARS-CoV-2 due to its distinct cytokine profile and the presence of burr cells in peripheral blood smears (130, 131).
MIS-C has been associated with heart failure in some cases (132).
A small number of case studies have identified presentations similar to MIS-C in adults associated with SARS-CoV-2 (133–136).
However, not all cases of severe COVID-19 in children are characterizable as MIS-C.
A recent study (137) described demographic and clinical variables associated with MIS-C in comparison with non-MIS-C severe acute COVID-19 in young people in the United States.
Efforts to characterize long-term sequelae of SARS-CoV-2 infection in children face the same challenges as in adults, but long-term effects remain a concern in pediatric patients (110, 138, 139), although some early studies have suggested that they may be less of a concern than in adults (140–142).
Research is ongoing into the differences between the pediatric and adult immune responses to SARS-CoV-2, and future research may shed light on the factors that lead to MIS-C; it is also unknown whether the relative advantages of children against severe COVID-19 will remain in the face of current and future variants (143).
1.5.2 Cytokine Release Syndrome
The inflammatory response was identified early on as a potential driver of COVID-19 outcomes due to existing research in SARS and emerging research in COVID-19.
While too low of an inflammatory response is a concern because it will fail to eliminate the immune threat (144), excessive pro-inflammatory cytokine activity can cascade (145) and cause cell damage, among other problems (146).
A dysregulated immune response can cause significant damage to the host (147–149) including pathogenesis associated with sepsis.
Sepsis, which can lead to multi-organ failure and death (150, 151), is traditionally associated with bacterial infections.
However, sepsis associated with viral infections may be underidentified (152), and sepsis has emerged as a major concern associated with SARS-CoV-2 infection (153).
Hyperactivity of the pro-inflammatory response due to lung infection is commonly associated with acute lung injury and more rarely with the more severe manifestation, ARDS, which can arise from pneumonia, SARS, and COVID-19 (145, 150).
Damage to the capillary endothelium can cause leaks that disrupt the balance between pro-inflammatory cytokines and their regulators (154), and heightened inflammation in the lungs can also serve as a source for systemic inflammation, or sepsis, and potentially multi-organ failure (150).
The shift from local to systemic inflammation is a phenomenon often referred to broadly as a cytokine storm (150) or, more precisely, as cytokine release syndrome (155).
Cytokine dysregulation is therefore a significant concern in the context of COVID-19.
In addition to the known role of cytokines in ARDS and lung infection more broadly, immunohistological analysis at autopsy of deceased SARS patients revealed that ACE2-expressing cells that were infected by SARS-CoV-1 showed elevated expression of the cytokines IL-6, IL-1β, and TNF-α (156).
Similarly, the introduction of the S protein from SARS-CoV-1 to mouse macrophages was found to increase production of IL-6 and TNF-α (157).
For SARS-CoV-2 infection leading to COVID-19, early reports described a cytokine storm syndrome-like response in patients with particularly severe infections (65, 158, 159).
Sepsis has been identified as a major contributor to COVID-19-related death.
Among patients hospitalized with COVID-19 in Wuhan, China, 112 out of 191 (59%) developed sepsis, including all 54 of the non-survivors (83).
While IL-6 is sometimes used as a biomarker for cytokine storm activity in sepsis (150), the relationship between cytokine profiles and the risks associated with sepsis may be more complex.
One study of patients with and at risk for ARDS, specifically those who were intubated for medical ventilation, found that shortly after the onset of ARDS, anti-inflammatory cytokine concentration in BALF increased relative to the concentration of pro-inflammatory cytokines (154).
The results suggest that an increase in pro-inflammatory cytokines such as IL-6 may signal the onset of ARDS, but recovery depends on an increased anti-inflammatory response (154).
However, patients with severe ARDS were excluded from this study.
Another analysis of over 1,400 pneumonia patients in the United States reported that IL-6, tumor necrosis factor (TNF), and IL-10 were elevated at intake in patients who developed severe sepsis and/or ultimately died (160).
However, unlike the study analyzing pro- and anti-inflammatory cytokines in ARDS patients (154), this study reported that unbalanced pro-/anti-inflammatory cytokine profiles were rare.
This discrepancy could be related to the fact that the sepsis study measured only three cytokines.
Although IL-6 has traditionally been considered pro-inflammatory, its pleiotropic effects via both classical and trans-signaling allow it to play an integral role in both the inflammatory and anti-inflammatory responses (161), leading it to be associated with both healthy and pathological responses to viral threat (162).
While the cytokine levels observed in COVID-19 patients fall outside of the normal range, they are not as high as typically found in patients with ARDS (163).
Regardless of variation in the anti-inflammatory response, prior work has therefore made it clear that pulmonary infection and injury are associated with systemic inflammation and with sepsis.
Inflammation has received significant interest both in regards to the pathology of COVID-19 as well as potential avenues for treatment, as the relationship between the cytokine storm and the pathophysiology of COVID-19 has led to the suggestion that a number of immunomodulatory pharmaceutical interventions could hold therapeutic value for the treatment of COVID-19 (3, 164).
1.6 Insights from Systems Biology
Systems biology provides a cross-disciplinary analytical paradigm through which the host response to an infection can be analyzed.
This field integrates the “omics” fields (genomics, transcriptomics, proteomics, metabolomics, etc.) using bioinformatics and other computational approaches.
Over the last decade, systems biology approaches have been used widely to study the pathogenesis of diverse types of life-threatening acute and chronic infectious diseases (165).
Omics-based studies have also provided meaningful information regarding host immune responses and surrogate protein markers in several viral, bacterial and protozoan infections (166).
Though the complex pathogenesis and clinical manifestations of SARS-CoV-2 infection are not yet fully understood, omics technologies offer the opportunity for discovery-driven analysis of biological changes associated with SARS-CoV-2 infection.
1.6.1 Transcriptomics
Through transcriptomic analysis, the effect of a viral infection on gene expression can be assessed.
Transcriptomic analyses, whether in vivo or in situ, can potentially reveal insights into viral pathogenesis by elucidating the host response to the virus.
For example, infection by some viruses, including by the coronaviruses SARS-CoV-2, SARS-CoV-1, and MERS-CoV, is associated with the upregulation of ACE2 in human embryonic kidney cells and human airway epithelial cells (65).
This finding suggests that SARS-CoV-2 facilitates the positive regulation of its own transmission between host cells (65).
The host immune response also likely plays a key role in mediating infection-associated pathologies.
Therefore, transcriptomics is one critical tool for characterizing the host response in order to gain insight into viral pathogenesis.
For this reason, the application of omics technologies to the process of characterizing the host response is expected to provide novel insights into how hosts respond to SARS-CoV-2 infection and how these changes might influence COVID-19 outcomes.
Several studies have examined the cellular response to SARS-CoV-2 in vitro in comparison to other viruses.
One study (167) compared the transcriptional responses of three human cell lines to SARS-CoV-2 and to other respiratory viruses, including MERS-CoV, SARS-CoV-1, Human parainfluenza virus 3, Respiratory syncytial virus, and Influenza A virus.
The transcriptional response differed between the SARS-CoV-1 infected cells and the cells infected by other viruses, with changes in differential expression specific to each infection type.
Where SARS-CoV-2 was able to replicate efficiently, differential expression analysis revealed that the transcriptional response was significantly different from the response to all of the other viruses tested.
A unique pro-inflammatory cytokine signature associated with SARS-CoV-2 was present in cells exposed to both high and low doses of the virus, with the cytokines IL-6 and IL1RA uniquely elevated in response to SARS-CoV-2 relative to other viruses.
However, one cell line showed significant IFN-I or IFN-III expression when exposed to high, but not low, doses of SARS-CoV-2, suggesting that IFN induction is dependent on the extent of exposure.
These results suggest that SARS-CoV-2 induces a limited antiviral state with low IFN-I or IFN-III expression and a moderate IFN-stimulated gene response, in contrast to other viruses.
Other respiratory viruses have been found to encode antagonists to the IFN response (168, 169), including SARS-CoV-1 (170) and MERS-CoV (171).
The analysis of SARS-CoV-2 suggested that this transcriptional state was specific to cells expressing ACE2, as it was not observed in cells lacking expression of this protein except with ACE2 supplementation and at very high (10-fold increase) level of SARS-CoV-2 exposure (167).
In another study, direct stimulation with inflammatory cytokines such as type I interferons (e.g., IFNβ) was also associated with the upregulation of ACE2 in human bronchial epithelial cells, with treated groups showing four-fold higher ACE2 expression than control groups at 18 hours post-treatment (172).
This hypothesis was further supported by studies showing that several nsp in SARS-CoV-2 suppress interferon activity (173) and that the SARS-CoV-2 ORF3b gene suppresses IFNB1 promoter activity (IFN-I induction) more efficiently than the SARS-CoV-1 ORF3b gene (174).
Taken together, these findings suggest that a unique cytokine profile is associated with the response to the SARS-CoV-2 virus, and that this response differs depending on the magnitude of exposure.
Susceptibility and IFN induction may also vary by cell type.
Using poly(A) bulk RNA-seq to analyzed dynamic transcriptional responses to SARS-CoV-2 and SARS-CoV-1 revealed negligible susceptibility of cells from the H1299 line (< 0.08 viral read percentage of total reads) compared to those from the Caco-2 and Calu-3 lines (>10% of viral reads) (175).
This finding suggests that the risk of infection varies among cell types, and that cell type could influence which hosts are more or less susceptible.
Based on visual inspection of microscopy images alongside transcriptional profiling, the authors also showed distinct responses among the host cell lines evaluated (175).
In contrast to Caco-2, Calu-3 cells infected with SARS-CoV-2 showed signs of impaired growth and cell death at 24 hours post infection, as well as moderate IFN induction with a strong up-regulation of IFN-stimulated genes.
Interestingly, the results were similar to those reported in Calu-3 cells exposed to much higher levels of SARS-CoV-2 (167), as described above.
This finding suggests that IFN induction in Calu-3 cells is not dependent on the level of exposure, in contrast to A549-ACE2 cells.
The discrepancy could be explained by the observations that Calu-3 cells are highly susceptible to SARS-CoV-2 and show rapid viral replication (34), whereas A549 cells are incompatible with SARS-CoV-2 infection (176).
This discrepancy raises the concern that in vitro models may vary in their similarity to the human response, underscoring the importance of follow-up studies in additional models.
As a result, transcriptional analysis of patient tissue is an important application of omics technology to understanding COVID-19.
Several studies have collected blood samples from COVID-19 patients and analyzed them using RNA-Seq (177–182).
Analyzing gene expression in the blood is valuable to understanding host-pathogen interactions because of the potential to identify alterations associated with the immune response and to gain insights into inflammation, among other potential insights (177).
One study compared gene expression in 39 COVID-19 inpatients admitted with community-acquired pneumonia to that of control donors using whole blood cell transcriptomes (177).
They also evaluated the effect of mild versus severe disease.
A greater number of differentially expressed genes were found in severe patients compared to controls than in mild patients compared to controls.
They also identified that the transcriptional profiles clustered into five groups and that the groups could not be explained by disease severity.
Most severe cases fell into two clusters associated with increased inflammation and granulocyte and neutrophil activation.
The presence of these clusters suggests the possibility that personalized medicine could be useful in the treatment of COVID-19 (177).
Longitudinal analysis of granulocytes from patients with mild versus severe COVID-19 revealed that granulocyte activation-associated factors differentiated the disease states, with greater numbers of differentially expressed genes early in disease course (177).
This study therefore revealed distinct patterns associated with COVID-19 and identified genes and pathways associated with each cluster.
Many other studies have also identified transcriptomic signatures associated with the immune response and inflammation.
Other studies have profiled the transcriptome of BALF (179) and the nasopharynx (183).
One study used single-cell transcriptomics techniques to investigate cell types including brain and choroid plexus cells compared to healthy controls and controls with influenza; among other signals of neuroinflammation, this study reported cortical T cells only in COVID-19 patients (184).
Transcriptomic analysis can thus provide insight into the pathogenesis of SARS-CoV-2 and may also be useful in identifying candidate therapeutics (177).
1.6.2 Proteomics
Proteomics analysis offers an opportunity to characterize the response to a pathogen at a level above transcriptomics.
Especially early on, this primarily involved evaluating the effect of the virus on cell lines.
One early proteomics study investigated changes associated with in vitro SARS-CoV-2 infection using Caco-2 cells (185).
This study reported that SARS-CoV-2 induced alterations in multiple vital physiological pathways, including translation, splicing, carbon metabolism and nucleic acid metabolism in the host cells.
Another area of interest is whether SARS-CoV-2 is likely to induce similar changes to other HCoV.
For example, because of the high level of sequence homology between SARS-CoV-2 and SARS-CoV-1, it has been hypothesized that sera from convalescent SARS-CoV-1 patients might show some efficacy in cross-neutralizing SARS-CoV-2-S-driven entry (33).
However, despite the high level of sequence homology, certain protein structures might be immunologically distinct, which would be likely to prohibit effective cross-neutralization across different SARS species (186).
Consequently, proteomic analyses of SARS-CoV-1 might also provide some essential information regarding the new pathogen (187, 188).
Proteomics research has been able to get ahead of the timeline for development of omics-level big data sets specific to SARS-CoV-2 by adopting a comparative bioinformatics approach.
Data hubs such as UniProt (189), NCBI Genome Database (190), The Immune Epitope Database and Analysis Resource (191), and The Virus Pathogen Resource (192) contain a wealth of data from studies in other viruses and even HCoV.
Such databases facilitate the systems-level reconstruction of protein-protein interaction networks, providing opportunities to generate hypotheses about the mechanism of action of SARS-CoV-2 and identify potential drug targets.
In an initial study (193), 26 of the 29 SARS-CoV-2 proteins were cloned and expressed in HEK293T kidney cells, allowing for the identification of 332 high-confidence human proteins interacting with them.
Notably, this study suggested that SARS-CoV-2 interacts with innate immunity pathways.
Ranking pathogens by the similarity between their interactomes and that of SARS-CoV-2 suggested West Nile virus, Mycobacterium tuberculosis, and human papillomavirus infections as the top three hits.
The fact that the host-pathogen interactome of the bacterium Mycobacterium tuberculosis was found to be similar to that of SARS-CoV-2 suggests that changes related to lung pathology might comprise a significant contributor to these expression profiles.
Additionally, it was suggested that the envelope protein, E, could disrupt host bromodomain-containing proteins, i.e., BRD2 and BRD4, that bind to histones, and the spike protein could likely intervene in viral fusion by modulating the GOLGA7-ZDHHC5 acyl-transferase complex to increase palmitoylation, which is a post-translational modification that affects how proteins interact with membranes (194).
An example of an application of this in silico approach comes from another study (195), which used patient-derived peripheral blood mononuclear cells to identify 251 host proteins targeted by SARS-CoV-2.
This study also reported that more than 200 host proteins were disrupted following infection.
In particular, a network analysis showed that nsp9 and nsp10 interacted with NF-Kappa-B-Repressing Factor, which encodes a transcriptional repressor that mediates repression of genes responsive to Nuclear Factor kappa-light-chain-enhancer of activated B-cells.
These genes are important to pro-, and potentially also anti-, inflammatory signaling (196).
This finding could explain the exacerbation of the immune response that shapes the pathology and the high cytokine levels characteristic of COVID-19, possibly due to the chemotaxis of neutrophils mediated by IL-8 and IL-6.
Finally, it was suggested (197) that the E protein of both SARS-CoV-1 and SARS-CoV-2 has a conserved Bcl-2 Homology 3-like motif, which could inhibit anti-apoptosis proteins, e.g., BCL2, and trigger the apoptosis of T cells.
Several compounds are known to disrupt the host-pathogen protein interactome, largely through the inhibition of host proteins.
Therefore, this research identifies candidate targets for intervention and suggests that drugs modulating protein-level interactions between virus and host could be relevant to treating COVID-19.
As with other approaches, analyzing the patterns found in infected versus healthy human subjects is also important.
COVID-19 infection has been associated with quantitative changes in transcripts, proteins, metabolites, and lipids in patient blood samples (198).
One longitudinal study (199) compared COVID-19 patients to symptomatic controls who were PCR-negative for SARS-CoV-2.
The longitudinal nature of this study allowed it to account for differences in the scale of inter- versus intraindividual changes.
At the time of first sampling, common functions of proteins upregulated in COVID-19 patients relative to controls were related to immune system mediation, coagulation, lipid homeostasis, and protease inhibition.
They compared these data to the patient-specific timepoints associated with the highest levels of SARS-CoV-2 antibodies and found that the actin-binding protein gelsolin, which is involved in recovery from disease, showed the steepest decline between those two time points.
Immunoglobulins comprised the only proteins that were significantly different between the COVID-19 and control patients at both of these timepoints.
The most significantly downregulated proteins between these time points were related to inflammation, while the most significantly upregulated proteins were immunoglobulins.
Proteins related to coagulation also increased between the two timepoints.
The selection of a symptomatic control cohort rather than healthy comparisons also suggests that the results are more likely to highlight the response to SARS-CoV-2 and COVID-19 specifically, rather than to disease more broadly.
This study also compared the disease course in patients who ultimately survived to those who died and found that ITIH4, a protein associated with the inflammatory response to trauma, may be a biomarker useful to identifying patients at risk of death.
Thus, these results indicate the value of studying patients in a longitudinal manner over the disease course.
By revealing which genes are perturbed during SARS-CoV-2 infection, proteomics-based analyses can thus provide novel insights into host-virus interaction and serve to generate new avenues of investigation for therapeutics.
1.7 Viral Virulence
Like that of SARS-CoV-1, the entry of SARS-CoV-2 into host cells is mediated by interactions between the viral spike glycoprotein, S, and human ACE2 (hACE2) (25, 33, 200–205).
Differences in how the S proteins of the two viruses interact with hACE2 could partially account for the increased transmissibility of SARS-CoV-2.
Studies have reported conflicting binding constants for the S-hACE2 interaction, though they have agreed that the SARS-CoV-2 S protein binds with equal, if not greater, affinity than the SARS-CoV-1 S protein does (16, 25, 203).
The C-terminal domain of the SARS-CoV-2 S protein in particular was identified as the key region of the virus that interacts with hACE2, and the crystal structure of the C-terminal domain of the SARS-CoV-2 S protein in complex with hACE2 reveals stronger interaction and a higher affinity for receptor binding than that of SARS-CoV-1 (204).
Among the 14 key binding residues identified in the SARS-CoV-1 S protein, eight are conserved in SARS-CoV-2, and the remaining six are semi-conservatively substituted, potentially explaining variation in binding affinity (25, 203).
Studies of crystal structure have shown that the RBD of the SARS-CoV-2 S protein, like that of other coronaviruses, undergoes stochastic hinge-like movement that flips it from a “closed” conformation, in which key binding residues are hidden at the interface between protomers, to an “open” one (16, 25).
Spike proteins cleaved at the furin-like binding site are substantially more likely to take an open conformation (66%) than those that are uncleaved (17%) (206).
Because the RBD plays such a critical role in viral entry, blocking its interaction with ACE2 could represent a promising therapeutic approach.
Nevertheless, despite the high structural homology between the SARS-CoV-2 RBD and that of SARS-CoV-1, monoclonal antibodies targeting SARS-CoV-1 RBD failed to bind to SARS-CoV-2-RBD (16).
However, in early research, sera from convalescent SARS patients were found to inhibit SARS-CoV-2 viral entry in vitro, albeit with lower efficiency than it inhibited SARS-CoV-1 (33).
Comparative genomic analysis reveals that several regions of the coronavirus genome are likely critical to virulence.
The S1 domain of the spike protein, which contains the receptor binding motif, evolves more rapidly than the S2 domain (23, 24).
However, even within the S1 domain, some regions are more conserved than others, with the receptors in S1’s N-terminal domain (S1-NTD) evolving more rapidly than those in its C-terminal domain (S1-CTD) (24).
Both S1-NTD and S1-CTD are involved in receptor binding and can function as RBDs to bind proteins and sugars (23), but RBDs in the S1-NTD typically bind to sugars, while those in the S1-CTD recognize protein receptors (12).
Viral receptors show higher affinity with protein receptors than sugar receptors (12), which suggests that positive selection on or relaxed conservation of the S1-NTD might reduce the risk of a deleterious mutation that would prevent binding.
The SARS-CoV-2 S protein also contains an RRAR furin recognition site at the S1/S2 junction (16, 25), setting it apart from both bat coronavirus RaTG13, with which it shares 96% genome sequence identity, and SARS-CoV-1 (207).
Such furin cleavage sites are commonly found in highly virulent influenza viruses (208, 209).
The furin recognition site at the S1/S2 junction is likely to increase pathogenicity via destabilization of the spike protein during fusion to ACE2 and the facilitation of cell-cell adhesion (16, 25, 42, 206, 208, 209).
These factors may influence the virulence of SARS-CoV-2 relative to other beta coronaviruses.
Additionally, a major concern has been the emergence of SARS-CoV-2 variants with increased virulence.
The extent to which evolution within SARS-CoV-2 may affect pathogenesis is reviewed below.
1.8 Molecular Signatures, Transmission, and Variants of Concern
Genetic variation in SARS-CoV-2 has been used to elucidate patterns over time and space.
Many mutations are neutral in their effect and can be used to trace transmission patterns.
Such signatures within SARS-CoV-2 have provided insights during outbreak investigations (210–212).
Similar mutations observed in several patients may indicate that the patients belong to the same transmission group.
The tracking of SARS-CoV-2 mutations is recognized as an essential tool for controlling future outbreaks and tracing the path of the spread of SARS-CoV-2.
In the first months of the pandemic in early 2020, early genomic surveillance efforts in Guangdong, China revealed that local transmission rates were low and that most cases arising in the province were imported (213).
Since then, efforts have varied widely among countries: for example, the U.K. has coordinated a national database of viral genomes (214), but efforts to collect this type of data in the United States have been more limited (215).
Studies have applied phylogenetic analyses of viral genomes to determine the source of local COVID-19 outbreaks in Connecticut (USA), (216), the New York City area (USA) (217), and Iceland (218).
There has been an ongoing effort to collect SARS-CoV-2 genomes throughout the COVID-19 outbreak, and as of summer 2021, millions of genome sequences have been collected from patients.
The sequencing data can be found at GISAID (219), NCBI (220), and COVID-19 data portal (221).
Ongoing evolution can be observed in genomic data collected through molecular surveillance efforts.
In some cases, mutations can produce functional changes that can impact pathogenesis.
One early example is the spike protein mutation D614G, which appeared in March 2020 and became dominant worldwide by the end of May 2020 (222, 223).
This variant was associated with increased infectivity and increased viral load, but not with more severe disease outcomes (222, 224).
This increased virulence is likely achieved by altering the conformation of the S1 domain to facilitate binding to ACE2 (224).
Similarly, the N439K mutation within the RBD of the spike protein is likely associated with increased transmissibility and enhanced binding affinity for hACE2, although it is also not thought to affect disease outcomes (225).
In contrast, a mutation in ORF8 that was identified in Singapore in the early months of 2020 was associated with cases of COVID-19 that were less likely to require treatment with supplemental oxygen (226), and a deletion surrounding the furin site insertion at the S1/S2 boundary has been identified only rarely in clinical settings (227), suggesting that these mutations may disadvantage viral pathogenesis in human hosts.
Thus, mutations have been associated with both virological and clinical differences in pathogenesis.
Several VOCs have also been identified and designated through molecular surveillance efforts (228).
The Alpha variant (lineage B.1.1.7) was first observed in the U.K. in October 2020 before it quickly spread around the world (229).
Other variants meriting further investigation have also been identified, including the Beta variant (B.1.351 lineage) first identified in South Africa and the Gamma variant (P.1 lineage) initially associated with outbreaks in Brazil.
These lineages share independently acquired mutations that may affect pathogenicity (230–234).
For example, they are all associated with a greater binding affinity for hACE2 than that of the wildtype variant (232, 235, 236), but they were not found to have more efficient cell entry than the wildtype virus (237).
A fourth VOC, the Delta variant (B.1.617.2 and AY.1, AY.2, and AY.3 lineages), was identified in India in late 2020 (238).
Some of the mutations associated with this lineage may alter fusogenicity and enhance furin cleavage, among other effects associated with increased pathogenicity (239).
The changes in these VOC demonstrate how ongoing evolution in SARS-CoV-2 can drive changes in how the virus interacts with host cells.
1.9 Quantifying Viral Presence
Assessing whether a virus is present in a sample is a more complex task than it initially seems.
Many diagnostic tests rely on real-time polymerase chain reaction (RT-PCR) to test for the presence versus absence of a virus (7).
They may report the cycle threshold (Ct) indicating the number of doubling cycles required for the target (in this case, SARS-CoV-2) to become detectable.
A lower Ct therefore corresponds to a higher viral load.
The Ct that corresponds to a positive can vary widely, but is often around 35.
This information is sufficient to answer many questions, since an amplicon must be present in order to be duplicated in RT-PCR.
For example, if a patient is presenting with COVID-19 symptoms, a positive RT-PCR test can confirm the diagnosis.
However, RT-PCR analysis alone cannot provide the information needed to determine whether a virus is present at sufficient levels to be infectious (240).
Some studies have therefore taken the additional step of cultivating samples in vitro in order to observe whether cells become infected with SARS-CoV-2.
One study collected upper respiratory tract samples from COVID-19 patients, analyzed them with RT-PCR to determine the cycle threshold, and then attempted to cultivate the SARS-CoV-2 virus in VeroE6 cells (240).
This study found that out of 246 samples, less than half (103) produced a positive culture.
Moreover, at a Ct of 35, only 5 out of 60 samples grew in vitro.
Therefore, the RT-PCR-confirmed presence of SARS-CoV-2 in a sample does not necessarily indicate that the virus is present at a high enough concentration to grow and/or spread.
1.10 Mechanisms of Transmission
When a human host is infected with a virus and is contagious, person-to-person viral transmission can occur through several possible mechanisms.
When a contagious individual sneezes, coughs, or exhales, they produce respiratory droplets that can contain a large number of viral particles (241).
Viral particles can enter the body of a new host when they then come in contact with the oral, nasal, eye, or other mucus membranes (241).
The primary terms typically used to discuss the transmission of viruses via respiratory droplets are droplet, aerosol, and contact transmission (242).
The distinction between droplet and aerosol transmission is typically anchored on whether a particle containing the virus is larger or smaller than 5 micrometers (μm) (243, 244).
Droplet transmission typically refers to contact with large droplets that fall quickly to the ground at close range, such as breathing in droplets produced by a sneeze (241, 243).
Aerosol transmission typically refers to much smaller particles (less than 5 μm) produced by sneezing, coughing, or exhaling (241, 242) that can remain suspended over a longer period of time and potentially to be moved by air currents (241).
It is also possible that viral particles deposited on surfaces via large respiratory droplets could later be aerosolized (241).
The transmission of viral particles that have settled on a surface is typically referred to as contact or fomite transmission (241, 245).
Any respiratory droplets that settle on a surface could contribute to fomite transmission (241).
Droplet and contact transmission are both well-accepted modes of transmission for many viruses associated with common human illnesses, including influenza and rhinovirus (241).
The extent to which aerosol transmission contributes to the spread of respiratory viruses is more widely debated.
In influenza A, for example, viral particles can be detected in aerosols produced by infected individuals, but it is not clear to what extent these particles drive the spread of influenza A infection (241, 242, 246–248).
Regardless of its role in the spread of influenza A, however, aerosol transmission likely played a role in outbreaks such as the 1918 Spanish Influenza (H1N1) and 2009 “swine flu” (pH1N1) (248).
All three of these mechanisms have been identified as possible contributors to the transmission of HCoVs (241), including the highly pathogenic coronaviruses SARS-CoV-1 and MERS-CoV (249, 250).
Transmission of SARS-CoV-1 is thought to proceed primarily through droplet transmission, but aerosol transmission is also considered possible (241, 251, 252), and fomite transmission may have also played an important role in some outbreaks (253).
Similarly, the primary mechanism of MERS transmission is thought to be droplets because inter-individual transmission appears to be associated with close interpersonal contact (e.g., household or healthcare settings), but aerosolized particles of the MERS virus have been reported to persist much more robustly than influenza A under a range of environmental conditions (254, 255).
However, few of these analyses have sought to grow positive samples in culture and thus to confirm their potential to infect new hosts.
Contact, droplet, and aerosol transmission are therefore all worth evaluating when considering possible modes of transmission for a respiratory virus like SARS-CoV-2.
The stability of the SARS-CoV-2 virus both in aerosols and on a variety of surfaces was found to be similar to that of SARS-CoV-1 (256).
Droplet-based and contact transmission were initially put forward as the greatest concern for the spread of SARS-CoV-2 (257), with droplet transmission considered the dominant mechanism driving the spread of the virus (258) because the risk of fomite transmission under real-world conditions is likely to be substantially lower than the conditions used for experimental analyses (259).
The COVID-19 pandemic has, however, exposed significant discrepancies in how terms pertaining to airborne viral particles are interpreted in different contexts (243).
The 5-μm distinction between “droplets” and “aerosols” is typical in the biological literature but is likely an artifact of historical science rather than a meaningful boundary in biology or physics (244).
Additionally, various ambient conditions such as air flow can influence how particles of different sizes fall or spread (243).
Despite initial skepticism about airborne transmission of SARS-CoV-2 through small particles (244), evidence now suggests that small particles can contribute to SARS-CoV-2 transmission (256, 260–262).
For example, one early study detected SARS-CoV-2 viral particles in air samples taken from hospitals treating COVID-19 patients, although the infectivity of these samples was not assessed (263).
Subsequently, other studies have been successful in growing SARS-CoV-2 in culture with samples taken from the air (264, 265) while others have not (266, 267) (see (268) for a systematic review of available findings as of July 2020).
The fact that viable SARS-CoV-2 may exist in aerosolized particles calls into question whether some axioms of COVID-19 prevention, such as 2-meter social distancing, are sufficient (244, 264, 269).
1.10.1 Symptoms and Viral Spread
Other aspects of pathogenesis are also important to understanding how the virus spreads, especially the relationship between symptoms, viral shedding, and contagiousness.
Symptoms associated with reported cases of COVID-19 range from mild to severe (8), but some individuals who contract COVID-19 remain asymptomatic throughout the duration of the illness (270).
The incubation period, or the time period between exposure and the onset of symptoms, has been estimated at five to eight days, with means of 4.91 (95% confidence interval (CI) 4.35-5.69) and 7.54 (95% CI 6.76-8.56) reported in two different Asian cities and a median of 5 (IQR 1 to 6) reported in a small number of patients in a Beijing hospital (271, 272).
However, the exact relationship between contagiousness and viral shedding remains unclear.
Estimates suggest that viral shedding can, in some cases, begin as early as 12.3 days (95% CI 5.9-17.0) before the onset of symptoms, although this was found to be very rare, with less than 0.1% of transmission events occurring 7 or more days before symptom onset (273).
Transmissibility appeared to peak around the onset of symptoms (95% CI -0.9 - 0.9 days), and only 44% (95% CI 30–57%) of transmission events were estimated to occur from presymptomatic contacts (273).
A peak in viral load corresponding to the onset of symptoms was also confirmed by another study (240).
As these trends became apparent, concerns arose due to the potential for individuals who did not yet show symptoms to transmit the virus (274).
Recovered individuals may also be able to transmit the virus after their symptoms cease.
A study of the communicable period based on twenty-four individuals who tested positive for SARS-CoV-2 prior to or without developing symptoms estimated that individuals may be contagious for one to twenty-one days, but they note that this estimate may be low (270).
In an early study, viral nucleic acids were reported to remain at observable levels in the respiratory specimens of recovering hospitalized COVID-19 patients for a median of 20 days and with a maximum observed duration through 37 days, when data collection for the study ceased (83).
As more estimates of the duration of viral shedding were released, they converged around approximately three weeks from first positive PCR test and/or onset of symptoms (which, if present, are usually identified within three days of the initial PCR test).
For example, in some studies, viral shedding was reported for up to 28 days following symptom onset (275) and for one to 24 days from first positive PCR test, with a median of 12 days (72).
On the other hand, almost 70% of patients were reported to still have symptoms at the time that viral shedding ceased, although all symptoms reduced in prevalence between onset and cessation of viral shedding (276).
The median time that elapsed between the onset of symptoms and cessation of viral RNA shedding was 23 days and between first positive PCR test and cessation of viral shedding was 17 days (276).
The fact that this study reported symptom onset to predate the first positive PCR test by an average of three days, however, suggests that there may be some methodological differences between it and related studies.
Furthermore, an analysis of residents of a nursing home with a known SARS-CoV-2 case measured similar viral load in residents who were asymptomatic regardless of whether they later developed symptoms, and the load in the asymptomatic residents was comparable to that of residents who displayed either typical or atypical symptoms (277).
Taken together, these results suggest that the presence or absence of symptoms are not reliable predictors of viral shedding or of SARS-CoV-2 status (e.g, (278)).
However, it should be noted that viral shedding is not necessarily a robust indicator of contagiousness.
The risk of spreading the infection was low after ten days from the onset of symptoms, as viral load in sputum was found to be unlikely to pose a significant risk based on efforts to culture samples in vitro(275).
The relationship between symptoms, detectable levels of the virus, and risk of viral spread is therefore complex.
The extent to which asymptomatic or presymptomatic individuals are able to transmit SARS-CoV-2 has been a question of high scientific and community interest.
Early reports (February and March 2020) described transmission from presymptomatic SARS-CoV-2-positive individuals to close family contacts (279, 280).
One of these reports (280) also included a description of an individual who tested positive for SARS-CoV-2 but never developed symptoms.
Later analyses also sought to estimate the proportion of infections that could be traced back to a presymptomatic or asymptomatic individual (e.g., (281)).
Estimates of the proportion of individuals with asymptomatic infections have varied widely.
The proportion of asymptomatic individuals on board the Diamond Princess cruise ship, which was the site of an early COVID-19 outbreak, was estimated at 17.9% (282).
In contrast, a model using the prevalence of antibodies among residents of Wuhan, China estimated a much higher rate of asymptomatic cases, at approximately 7 in 8, or 87.5% (283).
An analysis of the populations of care homes in London found that, among the residents (median age 85), the rate of asymptomatic infection was 43.8%, and among the caretakers (median age 47), the rate was 49.1% (284).
The duration of viral shedding may also be longer in individuals with asymptomatic cases of COVID-19 compared to those who do show symptoms (285).
As a result, the potential for individuals who do not know they have COVID-19 to spread the virus raises significant concerns.
In Singapore and Tianjin, two cities studied to estimate incubation period, an estimated 40-50% and 60-80% of cases, respectively, were considered to be caused by contact with asymptomatic individuals (271).
An analysis of viral spread in the Italian town of Vo’, which was the site of an early COVID-19 outbreak, revealed that 42.5% of cases were asymptomatic and that the rate was similar across age groups (286).
The argument was thus made that the town’s lockdown was imperative for controlling the spread of COVID-19 because it isolated asymptomatic individuals.
While more models are likely to emerge to better explore the effect of asymptomatic individuals on SARS-CoV-2 transmission, these results suggest that strategies for identifying and containing asymptomatic but contagious individuals are important for managing community spread.
1.10.2 Estimating the Fatality Rate
Estimating the occurrence of asymptomatic and mild COVID-19 cases is important to identifying the mortality rate associated with COVID-19.
The mortality rate of greatest interest would be the total number of fatalities as a fraction of the total number of people infected.
One commonly reported metric is the case fatality rate (CFR), which compares the number of COVID-19 related deaths to the number of confirmed or suspected cases.
However, in locations without universal testing protocols, it is impossible to identify all infected individuals because so many asymptomatic or mild cases go undetected.
Therefore, a more informative metric is the infection fatality rate (IFR), which compares the known deaths to the estimated number of cases.
It thus requires the same numerator as CFR, but divides by an approximation of the total number of cases rather than only the observed/suspected cases.
IFR varies regionally, with some locations observed to have IFRs as low as 0.17% while others are as high as 1.7% (287).
Estimates of CFR at the national and continental level and IFR at the continent level is maintained by the Centre for Evidence-Based Medicine (288).
Several meta-analyses have also sought to estimate IFR at the global scale.
These estimates have varied; one peer-reviewed study aggregated data from 24 other studies and estimated IFR at 0.68% (95% CI 0.53%–0.82%), but a preprint that aggregated data from 139 countries calculated a global IFR of 1.04% (95% CI 0.77%-1.38%) when false negatives were considered in the model (287, 289).
A similar prevalence estimate was identified through a repeated cross-sectional serosurvey conducted in New York City that estimated the IFR as 0.97% (290).
Examination of serosurvey-based estimates of IFR identified convergence on a global IFR estimate of 0.60% (95% CI 0.42%–0.77%) (287).
All of these studies note that IFR varies widely by location, and it is also expected to vary with demographic and health-related variables such as age, sex, prevalence of comorbidities, and access to healthcare and testing (291).
Estimates of infection rates are becoming more feasible as more data becomes available for modeling and will be bolstered as serological testing becomes more common and more widely available.
However, this research may be complicated due to the emergence of variants over time, as well as the varying availability and acceptance of vaccines in different communities and locations.
1.11 Dynamics of Transmission
Disease spread dynamics can be estimated using R0, the basic reproduction number, and Rt, the effective reproduction number.
Accurate estimates of both are crucial to understanding the dynamics of infection and to predicting the effects of different interventions.
R0 is the average number of new (secondary) infections caused by one infected person, assuming a wholly susceptible population (292), and is one of the most important epidemiological parameters (293).
A simple mechanistic model used to describe infectious disease dynamics is a susceptible-infected-recovered compartmental model (294, 295).
In this model, individuals move through three states: susceptible, infected, and recovered; two parameters, \(\gamma\) and \(\beta\), specify the rate at which the infectious recover, and the infection transmission rate, respectively, and R0 is estimated as the ratio of \(\beta\) and \(\gamma\)(293, 296).
A pathogen can invade a susceptible population only if R0 > 1 (293, 297).
The spread of an infectious disease at a particular time t can be quantified by Rt, the effective reproduction number, which assumes that part of the population has already recovered (and thus gained immunity to reinfection) or that mitigating interventions have been put into place.
For example, if only a fraction St of the population is still susceptible, Rt = St x R0.
When Rt is greater than 1, an epidemic grows (i.e., the proportion of the population that is infectious increases); when Rt is less than 1, the proportion of the population that is infectious decreases.
R0 and Rt can be estimated directly from epidemiological data or inferred using susceptible-infected-recovered-type models.
To capture the dynamics of SARS-CoV-2 accurately, the addition of a fourth compartment, i.e. a susceptible-exposed-infectious-recovered model, may be appropriate because such models account for the relative lengths of incubation and infectious periods (298).
Original estimates of R0 for COVID-19 lie in the range R0=1.4-6.5 (299–301).
Variation in R0 is expected between different populations, and the estimated values of R0 discussed below are for specific populations in specific environments.
The different estimates of R0 should not necessarily be interpreted as a range of estimates of the same underlying parameter.
In one study of international cases, the predicted value was R0=1.7 (302).
In China (both Hubei province and nationwide), the value was predicted to lie in the range R0=2.0-3.6 (299, 303, 304).
Another estimate based on a cruise ship where an outbreak occurred predicted R0=2.28 (305).
Susceptible-exposed-infectious-recovered model-derived estimates of R0 range from 2.0 - 6.5 in China (306–309) to R0=4.8 in France (310).
Using the same model as for the French population, a study estimated R0=2.6 in South Korea (310), which is consistent with other studies (311).
From a meta-analysis of studies estimating R0, (300) the median R0 was estimated to be 2.79 (IQR 1.16) based on twelve studies published between January 1 and February 7, 2020.
Inference of the effective reproduction number can provide insight into how populations respond to an infection and the effectiveness of interventions.
In China, Rt was predicted to lie in the range 1.6-2.6 in January 2020, before travel restrictions (312).
Rt decreased from 2.35 one week before travel restrictions were imposed (January 23, 2020), to 1.05 one week after.
Using their model, the authors also estimated the probability of new outbreaks occurring.
Assuming individual-level variation in transmission comparable to that of MERS or SARS, the probability of a single individual exporting the virus and causing a large outbreak is 17-25%, and assuming variation like that of SARS and transmission patterns like those observed for COVID-19 in Wuhan, the probability of a large outbreak occurring after ≥4 infections exist at a new location is greater than 50%.
An independent study came to similar conclusions, finding Rt=2.38 in the two-week period before January 23 with a decrease to Rt = 1.34 (using data from January 24 to February 3) or Rt=0.98 (using data from January 24 to February 8) (301).
In South Korea, Rt was inferred for February through March 2020 in two cities, Daegu (the center of the outbreak) and Seoul (311).
Metro data was also analyzed to estimate the effects of social distancing measures.
Rt decreased in Daegu from around 3 to <1 over the period that social distancing measures were introduced.
In Seoul, Rt decreased slightly, but remained close to 1 (and larger than Rt in Daegu).
These findings indicate that social distancing measures appeared to be effective in containing the infection in Daegu, but in Seoul, Rt remained above 1, meaning secondary outbreaks remained possible.
The study also shows the importance of region-specific analysis: the large decline in case load nationwide was mainly due to the Daegu region and could mask persistence of the epidemic in other regions, such as Seoul and Gyeonggi-do.
In Iran, estimates of Rt declined from 4.86 in the first week to 2.1 by the fourth week after the first cases were reported (313).
In Europe, analysis of 11 countries inferred the dynamics of Rt over a time range from the beginning of the outbreak until March 28, 2020, by which point most countries had implemented major interventions (such as stay-at-home orders, public gathering bans, and school closures) (314).
Across all countries, the mean Rt before interventions began was estimated as 3.87; Rt varied considerably, from below 3 in Norway to above 4.5 in Spain.
After interventions, Rt decreased by an average of 64% across all countries, with mean Rt=1.43.
The lowest predicted value was 0.97 for Norway and the highest was 2.64 for Sweden, which could be related to the fact that Sweden did not implement social distancing measures on the same scale as other countries.
The study concludes that while large changes in Rt are observed, it is too early to tell whether the interventions put into place are sufficient to decrease Rt below 1.
Evolution within SARS-CoV-2 has also driven changes in the estimated reproduction number for different populations at different times.
As of June 2021, the reproduction number had increased globally relative to 2020, and increased transmissibility over the wildtype variant was observed for the Alpha, Beta, Gamma, and Delta VOC (315).
In the U.S. between December 2020 and January 2021, B.1.1.7 (Alpha) was estimated to have an increased transmission of 35 to 45% relative to common SARS-CoV-2 variants at the time, with B.1.1.7 the dominant SARS-CoV-2 variant in some places at some timepoints (316).
This lineage was estimated to have increased transmissibility of 43 to 90% in the U.K. (317).
An estimate of the reproduction number of B.1.1.7 in the U.K. from September to December 2020 yielded 1.59 overall and between 1.56 and 1.95 in different regions of the country (234).
The Delta variant is particularly transmissible, and it has been estimated to be twice as transmissible than the wildtype variant of SARS-CoV-2 (315).
A review of the literature describing the Delta variant identified a mean estimated R0 of 5.08 (318).
Such differences can affect fitness and therefore influence the relative contributions of different lineages to a given viral gene pool over time (319).
Therefore, the evolution of the virus can result in shifts in the reproduction rate.
More generally, population-level epidemic dynamics can be both observed and modeled (296).
Data and empirically determined biological mechanisms inform models, while models can be used to try to understand data and systems of interest or to make predictions about possible future dynamics, such as the estimation of capacity needs (320) or the comparison of predicted outcomes among prevention and control strategies (321, 322).
Many current efforts to model Rt have also led to tools that assist the visualization of estimates in real time or over recent intervals (323, 324).
These are valuable resources, yet it is also important to note that the estimates arise from models containing many assumptions and are dependent on the quality of the data they use, which varies widely by region.
1.12 Effect of Vaccines on Pathogenesis and Community Spread
The vaccine clinical trial data demonstrate a significant reduction in the likelihood of contracting symptomatic COVID-19, thereby succeeding in the primary goal of vaccination.
The mRNA vaccines in particular were initially so effective in preventing disease that they were also assumed to have an effect on the likelihood of transmission (e.g., venues requiring proof of vaccination).
However, in light of the reduced efficacy in response to VOC, it is especially important to consider whether this assumption is supported by the available evidence.
This question is made up of several components.
The crux is whether vaccinated individuals with a SARS-CoV-2 infection, regardless of symptom status, are as contagious as unvaccinated, infected individuals.
Additionally, as outlined above, an important qualification is that the variants of SARS-CoV-2 circulating at the time of each study must be considered in light of the effect of evolution on vaccine efficacy.
The phase II/III clinical trials evaluating the mRNA vaccines assessed vaccine efficacy based on COVID-19 diagnosis, thereby detecting only patients who received a diagnosis.
In order to identify patients infected with SARS-CoV-2 who did not receive a diagnosis, for example, potentially those who did not develop symptoms, it would be necessary to conduct routine PCR testing even in the absence of symptoms.
Prior to the development of vaccines, the evidence suggested that asymptomatic individuals could spread SARS-CoV-2.
Investigation of viral dynamics of asymptomatic infection in early 2020 indicated that asymptomatic patients continued to shed the virus for a duration similar to that of symptomatic patients (325) (although viral shedding should not be conflated with contagiousness without further investigation).
Another study found viral load to be higher in the nasopharyngeal/oropharyngeal samples of asymptomatic patients compared to symptomatic patients hospitalized due to symptoms and/or known exposure (326).
However, the sample size in both of these studies was small, and a larger study found higher viral load in symptomatic than asymptomatic cases (327) along with a systematic review finding a reduced probability of asymptomatic transmission (328).
While far from conclusive, these studies suggest that asymptomatic cases still cary a risk of transmitting SARS-CoV-2.
One important consideration is therefore how likely vaccinated individuals are to develop asymptomatic SARS-CoV-2.
Considering asymptomatic cases is necessary to establish a more complete picture of efficacy with respect to spread.
Routine testing of healthcare workers in California who had received an mRNA vaccine revealed slightly higher rates of absolute risk for testing positive than those identified in the phase II/III trials, although the extent to which asymptomatic infection influenced these numbers was not investigated (329).
Another study analyzed the results of COVID-19 screening tests administered to asymptomatic individuals prior to receiving certain medical services at the Mayo Clinic in several locations across the United States.
This study found patients who had received two doses of an mRNA vaccine to be 73% less likely to have asymptomatic COVID-19 than patients who had received zero doses (330).
Because this study began on December 17, 2020, a date selected to coincide with the first day vaccines were available at the Mayo Clinic, this number may underestimate the efficacy of the vaccines given that many people eligible for early vaccination were at increased risk for exposure (e.g., healthcare workers and residents of long-term care facilities) (330).
In Israel, a longitudinal study of nearly 12,000 healthcare workers found that of the 5,372 fully vaccinated people with Pfizer/BioNTech BNT162b2, 8 developed symptomatic COVID-19 (0.15%) and 19 developed asymptomatic COVID-19 (0.35%) (331).
While the study itself analyzed the efficacy of the vaccine based on person-days, these findings also suggest that many or even the majority of SARS-CoV-2 infections in vaccinated individuals are likely to be asymptomatic.
Therefore, in addition to the symptomatic cases reported by the vaccine clinical trials, these findings suggest that asymptomatic cases can also occur in vaccinated people.
In the absence of symptoms, individuals are less likely to know to self-isolate, and therefore evaluating the effect of the vaccine on viral load is critical to understanding the role vaccinated individuals can play in spreading SARS-CoV-2.
Another question of interest is therefore whether vaccinated individuals positive for SARS-CoV-2 carry a similar viral load to unvaccinated individuals.
Viral load is often approximated by cycle threshold (Ct), or the cycle at which viral presence is detected during RT-qPCR, with a lower Ct corresponding to a greater viral load.
A prospective cohort study that evaluated front-line workers in six U.S. states from December 2020 to April 2021 reported a 40% reduction in viral load even with just a single dose of an mRNA vaccine (332).
The vaccine also appeared to influence the time to viral clearance: the risk of having detectable levels of SARS-CoV-2 for more than one week was reduced by 66% in participants who had received at least one dose (332).
However, this study compared the mean viral load across the two groups, meaning that these findings cannot be extrapolated across all points in the disease course.
Similarly, between December 2020 and February 2021, positive RT-qPCR tests were analyzed for almost 5,000 Israeli patients (333).
Ct was analyzed relative to when each patient received the first dose of the Pfizer mRNA vaccine.
A sharp increase in Ct (corresponding to reduced viral load) was observed between days 11 and 12, consistent with what is known about the onset of immunity following vaccination.
This pattern therefore suggested a direct effect of vaccination on viral load.
Other studies, however, have not offered support for a reduced viral load in breakthrough cases.
In Singapore, which has strict protocols for screening individuals with potential COVID-19 exposure, a retrospective cohort of patients who tested positive for SARS-CoV-2 between April and June 2021 was analyzed to compare viral kinetics and symptom course between vaccinated and unvaccinated cases.
Vaccinated individuals who tested positive experienced fewer symptoms than unvaccinated, SARS-CoV-2-positive individuals and were more likely to be asymptomatic (334).
Additionally, this study analyzed Ct over time and found that, though the median values were similar between the two groups at disease onset, viral load appeared to decrease more rapidly in vaccinated cases (334).
This study is likely to have evaluated a more accurate representation of all COVID-19 outcomes than has been feasible in most studies, but one limitation was that the RT-PCR reactions were conducted in many different facilities.
A third study investigated viral load (as approximated by Ct) using samples processed in a single laboratory during the summer of 2021 (335).
This study identified no significant differences in Ct between fully vaccinated and unvaccinated cases, but this study used samples sent for diagnosis and was not longitudinal.
It offered the additional benefit of culturing samples to assess whether their Ct threshold was likely to represent contagiousness and found that SARS-CoV-2 could be cultured from 51 of 55 samples with Ct less than 25 (the cut-off used in many studies).
Another study of samples collected at two sites in San Francisco, one of which tested only asymptomatic individuals, reported no difference in Ct between asymptomatic and symptomatic cases regardless of whether vaccination status was included in the model (336).
Though each of these three studies offers distinct strengths and weaknesses, taken together, they suggest that viral load is likely to be similar in vaccinated and unvaccinated individuals, but that vaccinated individuals clear the virus more rapidly, meaning that the average viral load is lower over time.
Given the emergence of VOC, especially the Delta and Omicron variants, for which breakthrough infections are more common, the potential for vaccinated individuals to spread SARS-CoV-2 is not static over time.
In fact, studies reporting reduced viral load in vaccinated individuals collected samples, for the most part, prior to the emergence of the Delta variant’s dominance.
The emergence of this variant may partially account for why more recent studies tend to find no difference between viral load in vaccinated and unvaccinated cases.
Taken together, these findings can provide some insight into how vaccines influence community spread.
While vaccinated individuals may be more likely to experience asymptomatic infection, current evidence about viral load in asymptomatic versus symptomatic cases is ambiguous.
Similarly, no conclusions can be drawn about whether viral load is different in vaccinated versus unvaccinated cases.
Therefore, at present, the evidence suggests that vaccinated individuals who are infected can still contribute to community spread.
The one potential mitigating factor supported at present is that differences in the viral kinetics may result in vaccinated cases infecting fewer individuals over time due to a more rapid decrease in viral load (334), although this study did not examine patterns in secondary transmission.
Thus, the virological evidence suggests that public health measures such as masking and distancing remain important even in areas with high vaccination rates.
1.13 Conclusions
The novel coronavirus SARS-CoV-2 is the third HCoV to emerge in the 21st century, and research into previous HCoVs has provided a strong foundation for characterizing the pathogenesis and transmission of SARS-CoV-2.
Critical insights into how the virus interacts with human cells have been gained from previous research into HCoVs and other viral infections.
With the emergence of three devastating HCoV over the past twenty years, emergent viruses are likely to represent an ongoing threat.
Contextualizing SARS-CoV-2 alongside other viruses serves not only to provide insights that can be immediately useful for combating this virus itself but may also prove valuable in the face of future viral threats.
Host-pathogen interactions provide a basis not only for understanding COVID-19, but also for developing a response.
As with other HCoVs, the immune response to SARS-CoV-2 is likely driven by detection of its spike protein, which allows it to enter cells through ACE2.
Epithelial cells have also emerged as the major cellular target of the virus, contextualizing the respiratory and gastrointestinal symptoms that are frequently observed in COVID-19.
Many of the mechanisms that facilitate the pathogenesis of SARS-CoV-2 are currently under consideration as possible targets for the treatment or prevention of COVID-19 (2, 3).
Research in other viruses also provides a foundation for understanding the transmission of SARS-CoV-2 among people and can therefore inform efforts to control the virus’s spread.
Airborne forms of transmission (droplet and aerosol transmission) have emerged as the primary modes by which the virus spreads to new hosts.
Asymptomatic transmission was also a concern in the SARS outbreak of 2002-03 and, as in the current pandemic, presented challenges for estimating rates of infection (337).
These insights are important for developing a public health response, such as the CDC’s shift in its recommendations surrounding masking (338).
Even with the background obtained from research in SARS and MERS, COVID-19 has revealed itself to be a complex and difficult-to-characterize disease that has many possible presentations that vary with age.
Variability in presentation, including cases with no respiratory symptoms or with no symptoms altogether, were also reported during the SARS epidemic at the beginning of the 21st century (337).
The variability of both which symptoms present and their severity have presented challenges for public health agencies seeking to provide clear recommendations regarding which symptoms indicate SARS-CoV-2 infection and should prompt isolation.
Asymptomatic cases add complexity both to efforts to estimate statistics such as R0 and Rt, which are critical to understanding the transmission of the virus, and IFR, which is an important component of understanding its impact on a given population.
The development of diagnostic technologies over the course of the pandemic has facilitated more accurate identification, including of asymptomatic cases (7).
As more cases have been diagnosed, the health conditions and patient characteristics associated with more severe infection have also become more clear, although there are likely to be significant sociocultural elements that also influence these outcomes (339).
While many efforts have focused on adults, and especially older adults because of the susceptibility of this demographic, additional research is needed to understand the presentation of COVID-19 and MIS-C in pediatric patients.
As more information is uncovered about the pathogenesis of HCoV and SARS-CoV-2 specifically, the diverse symptomatology of COVID-19 has and likely will continue to conform with the ever-broadening understanding of how SARS-CoV-2 functions within a human host.
While the SARS-CoV-2 virus is very similar to other HCoV in several ways, including in its genomic structure and the structure of the virus itself, there are also some differences that may account for differences in the COVID-19 pandemic compared to the SARS and MERS epidemics of the past two decades.
The R0 of SARS-CoV-2 has been estimated to be similar to SARS-CoV-1 but much higher than that of MERS-CoV (340), although a higher R0 has been estimated for some VOC.
While the structures of the viruses are very similar, evolution among these species may account for differences in their transmissibility and virulence.
For example, the acquisition of a furin cleavage site the S1/S2 boundary within the SARS-CoV-2 S protein may be associated with increased virulence.
Additionally, concerns have been raised about the accumulation of mutations within the SARS-CoV-2 species itself, and whether these could influence virulence (341).
These novel variants may be resistant to vaccines and antibody treatments such as Bamlanivimab that were designed based on the wildtype spike protein (3, 6).
As a consequence of reliance on targeting the SARS-CoV-2 spike protein for many therapeutic and prophylactic strategies, increased surveillance is required to rapidly identify and prevent the spread of novel SARS-CoV-2 variants with alterations to the spike protein.
The coming of age of genomic technologies has made these types of analyses feasible, and genomics research characterizing changes in SARS-CoV-2 along with temporal and spatial movement is likely to provide additional insights into whether within-species evolution influences the effect of the virus on the human host.
Additionally, the rapid development of sequencing technologies over the past decade has made it possible to rapidly characterize the host response to the virus.
For example, proteomics analysis of patient-derived cells revealed candidate genes whose regulation is altered by SARS-CoV-2 infection, suggesting possible approaches for pharmaceutical invention and providing insight into which systems are likely to be disrupted in COVID-19 (195).
As more patient data becomes available, the biotechnological advances of the 2000s are expected to allow for more rapid identification of potential drug targets than was feasible during the SARS, or even MERS, pandemic.
Thus, the COVID-19 crisis continues to evolve, but the insights acquired over the past 20 years of HCoV research have provided a solid foundation for understanding the SARS-CoV-2 virus and the disease it causes.
As the scientific community continues to respond to COVID-19 and to elucidate more of the relationships between pathogenesis, transmission, host regulatory responses, and symptomatology, this understanding will no doubt continue to evolve and to reveal additional connections among virology, pathogenesis, and health.
This review represents a collaboration between scientists from diverse backgrounds to contextualize this virus at the union of many different biological disciplines (4).
At present, understanding the SARS-CoV-2 virus and its pathogenesis is critical to a holistic understanding of the COVID-19 pandemic.
In the future, interdisciplinary work on SARS-CoV-2 and COVID-19 may guide a response to a new viral threat.
2 Evolutionary Perspectives on SARS-CoV-2
2.1 Abstract
2.2 Importance
2.3 Introduction
The emergence of what is now known to be the pathogen SARS-CoV-2 has dramatically reshaped modern life for the past two years.
The genomic revolution provided the tools needed to understand the virus in ways that were not feasible during previous pandemics.
For example, the first genome sequence of the pathogen was released on January 3, 2020, providing valuable information about the pathogen within a month and a half of the first known cases.
As the pandemic has unfolded, evolutionary questions and methods of investigation have framed the scientific approach to understanding the virus.
These questions have evolved along with the pandemic.
Thus far, five major evolutionary questions have emerged.
The first was “what is it?”, the second “where did it come from?”, the third and fourth address “whom does it affect?”, the fifth “how is it changing?” and the sixth “what is next?”
Evolutionary biology provides a framework through which these questions can be evaluated and explored.
2.4 Question 1: What Is It?
What is now known as SARS-CoV-2 emerged in November 2019 as an unknown pathogen causing a cluster of pneumonia cases in Wuhan, China.
The initial genome sequence, which was released in early January 2020, revealed the pathogen to be a novel coronavirus (11).
Although most coronaviruses show little transmission in humans, several human coronaviruses (HCoV) have been identified since the 1960s.
Therefore, in the early days of the pandemic, many strategies to understand or manage the emergent viral threat focused on contextualizing it amongst better-studied coronaviruses.
Many people have previously been infected by an HCoV.
Approximately one-third of common cold infections are thought to be caused by four seasonal HCoV: Human coronavirus 229E (HCoV-229E), Human coronavirus NL63 (HCoV-NL63), Human coronavirus OC43 (HCoV-OC43), and Human coronavirus HKU1 (HCoV-HKU1) (342–344).
The first HCoV were identified in the 1960s: HCoV-229E in 1965 (345) and HCoV-OC43 in 1967 (346).
Both of these viruses typically cause cold-like symptoms, including upper and lower respiratory infections (347–349), but they have also been associated with gastrointestinal symptoms (350).
Two additional HCoV were subsequently identified (351, 352).
In 2003, HCoV-NL63 (351) was first identified in a 7-month-old infant and then in clinical specimens collected from seven additional patients, five of whom were infants younger than 1 year old and the remainder of whom were adults.
CoV-HKU1 was identified in samples collected from a 71-year-old pneumonia patient in 2004 and then found in samples collected from a second adult patient (352).
These viruses are associated with respiratory diseases of varying severity, ranging from common cold to severe pneumonia, with severe symptoms mostly observed in immunocompromised individuals (353), and also have gastrointestinal involvement in some cases (350).
In addition to these relatively mild HCoV, however, highly pathogenic human coronaviruses have been identified, including Severe acute respiratory syndrome-related coronavirus (SARS-CoV or SARS-CoV-1) and Middle East respiratory syndrome-related coronavirus (MERS-CoV) (250, 342, 354).
At the time that SARS-CoV-1 emerged in the early 2000s, no HCoV had been identified in almost 40 years (250).
The first case of SARS was reported in November 2002 in the Guangdong Province of China, and over the following month, the disease spread more widely within China and then into several countries across multiple continents (250, 340).
Unlike previously identified HCoV, SARS was much more severe, with an estimated death rate of 9.5% (340).
It was also highly contagious via droplet transmission, with a basic reproduction number (R0) of 4 (i.e., each person infected was estimated to infect four other people) (340).
However, the identity of the virus behind the infection remained unknown until April of 2003, when the SARS-CoV-1 virus was identified through a worldwide scientific effort spearheaded by the WHO (250).
SARS-CoV-1 belonged to a distinct lineage from the two other HCoV known at the time (340).
By July 2003, the SARS outbreak was officially determined to be under control, with the success credited to infection management practices (250).
A decade later, a second outbreak of severe respiratory illness associated with a coronavirus emerged, this time in the Arabian Peninsula.
This disease, known as Middle East respiratory syndrome (MERS), was linked to another novel coronavirus, MERS-CoV.
The fatality rate associated with MERS is much higher than that of SARS, at almost 35%, but the disease is much less easily transmitted, with an R0 of 1 (340).
Although MERS is still circulating, its low reproduction number has allowed for its spread to be contained (340).
The COVID-19 pandemic is thus associated with the seventh HCoV to be identified and the fifth since the turn of the millennium, though additional HCoVs may be in circulation but remain undetected (e.g., (355)).
Following the release of the SARS-CoV-2 genome sequence, multiple research groups sequenced the genomes of SARS-CoV-2 specimens identified in clinical samples.
These samples were primarily collected from patients’ lower respiratory tract, namely bronchoalveolar lavage fluid (BALF), and the upper respiratory tract, in the form of throat and nasopharyngeal swabs (19, 207, 356).
Integration of these sequences allowed for a more complete picture of the viral genome.
Analysis of the viral genome revealed significant sequence homology with two known HCoV: the novel coronavirus shared about 79% sequence identity with SARS-CoV-1 and 50% with MERS-CoV (19).
Therefore, this early phylogenetic analysis of the novel coronavirus allow its similarity to other, known viruses to be established.
SARS-CoV-1 and MERS-CoV were ultimately managed largely through infection management practices (e.g., mask wearing) and properties of the virus itself (i.e., low rate of transmission), respectively (250, 340).
Research in response to prior outbreaks of HCoV-borne infections, such as SARS and MERS, provided a strong foundation for hypotheses about the pathogenesis of SARS-CoV-2 as well as potential diagnostic and therapeutic approaches, as we review elsewhere (1, 3, 5, 6).
Therefore, this phylogenetic information was valuable for gaining an understanding of the pathogen and identifying strategies to manage it.
2.5 Question 2: Where Did It Come From?
Despite the high degree of similarity to SARS-CoV-1, even greater sequence identity was observed between SARS-CoV-2 and zoonotic coronaviruses.
A 2001 literature review estimated that 61% of human pathogens have a zoonotic origin (357).
A zoonotic disease, or zoonosis, arises when a pathogen can both a) infect and b) cause a disease in humans (358).
As a result, the risk of zoonotic disease increases when there is substantial interaction between humans and wildlife (358).
Many factors can influence this human/wildlife interface and therefore the risk of zoonotic transmission events (358, 359).
In the SARS epidemic, SARS-CoV-1 was also thought to have emerged in a live animal market.
A survey of a market in Shenzhen, China revealed that individuals from two carnivore species, namely several masked palm civets (Paguma larvata) and one raccoon dog (Nyctereutes procyonoides), were likely carriers of SARS-CoV-1, despite presenting as healthy (360).
However, further analysis suggested that these species might be only intermediate hosts who were exposed in the market setting (361).
A closely related virus was identified in Chinese horseshoe bats (Rhinolophus sinicus), but the sequence identity was only 88% with SARS-CoV-1 (362).
Therefore, the species of origin for SARS-CoV-1 remains unresolved.
In the case of SARS-CoV-2, early interest for the emergence of the pathogen turned to live-animal markets in Wuhan (363, 364–add-to-Wuhan-riddle), where it would later emerge that many animals were sold suffering from poor health and hygiene (365).
A large percentage of early patients had visited the Huanan seafood market in Wuhan, and next-generation sequencing of samples collected from nine patients, eight of whom had visited the market, revealed extremely high sequence identity (99.98%), indicative of rapid spread (19).
The sequence of the viral pathogen collected from these patients was also compared to known zoonotic pathogens.
In particular, genomic research quickly highlighted significant similarity (about 88% sequence identity) between SARS-CoV-2 and bat-derived SARS-like coronaviruses, namely bat-SL-CoVZC45 and bat-SL-CoVZXC21 (19).
Other analyses have reported even greater similarity between SARS-CoV-2 and the bat coronavirus BatCoV-RaTG13, with shared sequence identity as high as 96.2% (207, 211).
Bats are well-established as a disease reservoir, including for RNA viruses (366–368).
This evidence therefore suggested that the virus may have emerged as a result of zoonotic transfer of a virus from bats to humans, with the wildlife trade considered a potential source of exposure.
Nevertheless, some fragments of the genome differ between SARS-CoV-2 and RATG13 by up to 17%, suggesting a complex natural selection process.
Additionally, SARS-CoV-2 is closely related (91.02%) to a novel coronaviruses identified in Malayan pangolins (Manis javanica) infected with a respiratory disease in October 2019 (369).
Although the genome-wide sequence identity was lower between SARS-CoV-2 and this pangolin virus than BatCoV-RaTG13, its particularly high similarity in the receptor binding domain (RBD) of the spike (S) gene with SARS-CoV-2 drew further attention (369, 370).
The SARS-CoV-2 RBD differs from the pangolin coronavirus RBD by only one amino acid change (369), and the sequence identity between the regions is 97.4% (370).
Pangolins were therefore identified as a potential intermediate host of SARS-CoV-2 between bats and humans.
However, data collected from May 2017 to November 2019 by a research team interested in tick-borne illnesses identified no bats or pangolins sold at these markets leading up to the emergence of COVID-19 (365).
Additionally, endemic bat species are typically in hibernation at the time that SARS-CoV-2 emerged (19).
Therefore, it is possible that animals associated with these markets were infected by bats, but it is not clear whether the disease emerged in a different location and/or whether it is associated with a different species.
There were 38 species observed at the market in the 2.5 years leading up to the emergence of SARS-CoV-2, indicating significant diversity in the animals with which humans were interacting (365).
As with SARS-CoV-1, the species of origin for SARS-CoV-2 therefore remains unresolved.
Genomic analyses and comparisons to other known coronaviruses suggest that SARS-CoV-2 is unlikely to have originated in a laboratory – either purposely engineered and released, or escaped – and instead evolved naturally in an animal host (371).
However, potentially due to public misunderstanding about recombination and complex evolutionary processes like coevolution, the similarity to pangolin S has resulted in popular conspiracy theories that the virus did not arise naturally.
The similarity of S to that of pangolin viruses could arise from either recombination or coevolution (211, 372), rather than requiring human intervention.
Such suspicions may also have been fueled, in part, by the lack of well-characterized bat coronaviruses which means that SARS-CoV-2 is still relatively derived from documented coronaviruses surveyed in bats (373).
While it has been suggested that more thorough investigation of the origins of COVID-19 may have some value (374), in many cases, support for the “lab-leak” theory is politically motivated (375).
A more robust panel of zoonotic viruses against which to compare SARS-CoV-2 would allow for conclusive dismissal of these politicized claims, underscoring another potential benefit of more thorough monitoring of zoonotic diseases.
More importantly, it would allow researchers to have a better understanding of and to community concerns about potential emerging viral threats.
2.6 Question 3: Which Species Are Susceptible?
Given the strong evidence for a zoonotic origin of SARS-CoV-2, another evolutionary question that received significant attention, especially early on, was whether humans could infect other species with SARS-CoV-2.
In the modern age, opportunities for human-to-animal transmission events could arise in interactions with companion animals, zoo animals, house pests, hunting, urbanized wildlife, and livestock.
Outbreaks of zoonotic diseases have been known to originate in environments such as zoos, farms, and petting zoos (376), indicating that disease transmission is likely to be possible in these contexts.
Additionally, many coronaviruses infect animals and have been the subject of veterinary medical investigations and vaccine development efforts due to their effect on the health of companion and agricultural animals (377).
Concerns about anthroponotic (human-to-animal) transmission focused on a few issues.
First, if animal species were susceptible to COVID-19-like infection, in addition to concerns about animal health, infections in livestock could have significant effects on food supply chains.
Additionally, even if pathology in these species was limited, if they could serve as viral reservoirs, then they would pose additional risk to humans.
The breadth of species susceptible to infection by a pathogen is known as the pathogen’s host range (378).
Understanding the host-pathogen relationship throughout SARS-CoV-2’s host range can therefore offer valuable information for managing the spread of SARS-CoV-2.
The phylogeny of the species implicated in the origination of COVID-19 suggested that the host range of SARS-CoV-2 could encompass many species with a high level of interaction with humans.
Humans last shared an ancestor with bats and pangolins almost 100 million years ago (379).
Bats belong to the order Chiroptera and pangolins to Pholidota, which both belong to the clade Pegasoferae(380–382).
They are closely related to many other species that have close relationships with humans, namely odd-toed ungulates (Euungulata) and carnivores (Carnivora) (380–383).
The part of the evolutionary tree that includes both humans and the Pegasoferae encompasses many species of significant social and economic importance.
Therefore, concerns were raised that the species with which humans have close interactions, many of which are much more closely related to bats and pangolins than humans are, could also be infected.
It seemed plausible that the host range could include both livestock, many of which are odd-toed ungulates, and companion animals, many of which are carnivores.
Infection of these animals was identified as a major concern (384).
Genomic analyses seeking to identify which species were likely to be susceptible focused largely on the comparative genetics of angiotensin-converting enzyme 2 (ACE2).
ACE2 is the primary protein used by SARS-CoV-2 to enter the cell (see (1)).
Recognition of this protein is largely determined by domains in the S1 subunit of the RBD (25).
Alignment of the ACE2 sequence from 19 species revealed high conservation among mammals (385).
This analysis suggested that non-human primates (three monkey and two ape species), companion animals (dogs and cats), and livestock (both odd- and even-toed ungulates) may all be susceptible to SARS-CoV-2 (385).
Similarly, another study conducted an in silico analysis of ACE2 protein structures and their predicted binding to SARS-CoV-2 for 410 vertebrate species (386).
The species identified as having the highest predicted binding affinities were all primates, including humans.
Other taxa with high predicted affinities included other primates, rodents, even-toed ungulates (namely, several species of cetaceans and deer), and anteaters.
Reindeer were the only domesticated species predicted to belong to either of these groups, but many common zoo animal species with threatened or worse IUCN risk status were identified as at risk.
Considering the evidence generated by in silico studies, it may not be surprising that many cases of reverse zoonotic, or anthroponotic, SARS-CoV-2 transmission have been reported.
Ferrets (Mustela furo) as well as cats and dogs were reported to be susceptible to SARS-CoV-2 in an experimental infection study (387).
The earliest reported anthroponotic transmission events were observed in house pets, primarily cats (Felis catus) (388–390).
Similarly, cases of SARS-CoV-2 infection have been reported in dogs (Canis familiaris): two of fifteen dogs monitored for SARS-CoV-2 by the Hong Kong Agriculture, Fisheries, and Conservation Department during the owners’ quarantine in March 2020 were found to be positive for SARS-CoV-2 (389).
Comparing estimates in studies where cats (Felis catus) living with SARS-CoV-2-positive humans were tested for SARS-CoV-2 suggest that 6 to 15% of house cats may become infected (388), and a large-scale study of pet dogs and cats in Italy suggested that 4.5% of cats and 12.8% of dogs from known COVID-19-positive households had developed antibodies to the virus (391).
Some of these SARS-CoV-2-positive domestic carnivores have also shown clinical symptoms (392), and a pilot study of seven cats and three dogs found that cats, but not dogs, shed SARS-CoV-2 virus for several days after viral challenge, although none of the animals were symptomatic (393).
A few dogs and cats have reportedly died after becoming infected with SARS-CoV-2, although in most cases whether the virus is causally related to the death is unclear (394–396, 397/?sh=4b653381275e, 398, 399).
Domestic pests, on the other hand, seem to be less susceptible to SARS-CoV-2.
In the comparative genomic analysis of ACE2, the two rodent species analyzed, despite being the most phylogenetically similar to humans aside from the other primates, showed the most sequence divergence in ACE2(385).
This finding was supported by experimental evidence that SARS-CoV-2 cannot use mouse (Mus musculus) ACE2 for cell entry (207).
In fact, research using murine models to study SARS and COVID-19 therefore uses transgenic mice designed to be sensitive to the virus (as summarized in (400)).
Similarly, SARS-CoV-2 in livestock also raised concern because of the potential effect on food supply.
However, studies using in vivo viral challenge reported that livestock species in general do not develop clinical manifestations of SARS-CoV-2 and do not shed infectious virus (387, 401).
In vitro exposure to SARS-CoV-2 suggested that sheep (Ovis aries), but not cattle (Bos taurus), might be susceptible to infection, but in vivo viral challenge suggested that sheep did not show notable susceptibility to infection (402).
Similarly, analyses of antibody response (403) suggested that sheep exposed to a high level of human interaction did not appear to have developed infections.
Following viral challenge of several species, including cattle, sheep, and horses (Equus ferus caballus), none were found to shed culturable levels of virus (401).
As a note, despite the low risk posed by livestock themselves, the working conditions of the meat industry itself were associated with a very high risk of SARS-CoV-2 infection for workers that did cause disruptions to food supply chains (404, 405).
However, one species of domesticated agricultural animal severely affected by SARS-CoV-2 was the mink (Neovison vison).
While fur farming has declined significantly since the twentieth century, mink farming is still common in China and some European countries, and mink farms continue to exist in the United States.
Mink belong to the Mustelidae family within Carnivora.
SARS-CoV-2 was first reported on mink farms in the Netherlands and Denmark in 2020 (406, 407).
Mink were observed to show symptoms of respiratory infection, with varied severity among individuals (406).
Dissection revealed lung pathology consistent with interstitial pneumonia (406).
An analysis of five farms in the United States reported mortality rates between 35 and 55% of adult minks (408).
Subsequently, mink farms worldwide reported outbreaks of SARS-CoV-2.
Concerns were amplified when novel variants of SARS-CoV-2 were identified as having emerged on Danish mink farms and spread into the human population (407, 407, 409–411).
The fact that these variants appeared in mink populations before being observed in humans suggests that mink can indeed serve as a viral reservoir (407).
Concerns about mink-to-human transmission led to the mass destruction of domesticated mink populations in Europe (412, 413).
Introgression from fur farms into wild populations (i.e., feralization) may have also resulted in the spread of SARS-CoV-2 into wild mink populations (414, 415).
Therefore, while the specific zoonotic origin of SARS-CoV-2 may still not be clear, the potential for the virus to take hold in species other than humans has been clearly demonstrated by the mink outbreak.
Finally, some species of zoo animals were also monitored to determine whether they were at risk.
Several species closely related to humans (i.e., the Great Apes) are threatened with extinction and had been identified through in silico studies as likely to be susceptible to SARS-CoV-2 (386), and therefore the potential for a virus to target these close relatives presented a major concern.
In early 2021, three gorillas (Gorilla beringei beringei) at the San Diego Zoo Safari Park developed respiratory symptoms that were confirmed to be associated with SARS-CoV-2 (416).
Gorillas at other zoos have also been infected (417–419).
Additionally, given the susceptibility of house cats, it is not so surprising that other felids are also susceptible to SARS-CoV-2.
Infections of several “big cats” including Malayan tigers (Panthera tigris jacksoni), Amur tigers (Panthera tigris altaica), and African lions (Panthera leo krugeri) were reported at New York City’s Bronx Zoo in March 2020 (420).
In late 2020, four lions (Panthera leo bleyenberghi) at the Barcelona Zoo also developed respiratory symptoms that were found to be caused by SARS-CoV-2 (411).
Several captive snow leopards (Panthera uncia) in the United States have died from COVID-19 (421, 422).
While discussions of zoonoses often focus on the risk that animal diseases carry for human populations, the COVID-19 pandemic has also underscored the risks that human diseases pose for animals.
COVID-19 precautions may have reduced the spread of other respiratory illnesses to wild mountain gorilla (Gorilla beringei beringei) populations (423), reducing one of the most significant threats to this endangered species (424).
In the case of gorillas, the potential for cross-species application of pharmaceutical advances has also become clear: captive gorillas with COVID-19 received monoclonal antibodies (425).
Additionally, several companies are developing veterinary vaccines against SARS-CoV-2.
The most visible has been Zoetis, a veterinary pharmaceutical company, that has developed vaccines that have been administered to several species, including felids in zoos, minks, and gorillas (426–429).
Russian researchers have also developed a COVID-19 vaccine for carnivores (430).
Therefore, the host range of SARS-CoV-2 is broad, including primates, bats, and carnivores.
The most severe infections have been observed in humans, felids, and mustelids (426).
In the United States, as of late 2021, dogs and cats made up the majority of non-human SARS-CoV-2 infections (431), but the most severe infections were observed in felids and mustelids (in addition to humans) (426).
Interestingly, comparing ACE2 binding activity across species (432, 433) revealed that it did not always align with which species known to be susceptible to SARS-CoV-2 infection, suggesting other binding sites might also be important.
While the specific zoonotic origins of SARS-CoV-2 remain unknown, pharmaceutical developments in the treatment of COVID-19 have included non-human species.
The complex relationship between animals, humans, and disease highlights the importance of a broad perspective on health that extends beyond a single species.
2.7 Question 4: Do Genes Influence Who is Affected?
Throughout the pandemic, many hypotheses have been raised about factors that might influence individuals’ susceptibility to COVID-19 or to severe disease .
Many risk factors, such as underlying health conditions, are related to the body’s inflammatory response, as we review elsewhere (339).
Here, we focus narrowly on genetic bases of differences in susceptibility or outcomes.
Historically, the identification of genetic risk factors for a disease typically utilized a candidate gene approach, where a gene of interest was evaluated to identify variants that showed an association with the outcome of interest.
While economical in terms of sequencing, this approach is prone to spurious results when applied to complex traits (434).
Today, in the age of next-generation sequencing (NGS), alternative approaches have emerged.
NGS makes it possible to conduct genome-wide scans where a large number of single-nucleotide polymorphisms (SNPs) or variants are evaluated to identify regions of the genome associated with variation in a phenotype.
Genome-wide association studies (GWAS) in particular are a popular approach that employs this strategy.
During COVID-19, both of these paradigms have been applied to the problem of identifying genetic correlates of disease severity.
2.7.1 Candidate-Gene Approaches
Many candidate genes have been investigated throughout the pandemic.
Here, we review three examples of candidate gene studies in COVID-19.
First, an early study (published in April 2020) investigated a known variant in interferon-induced transmembrane protein 3 (IFITM3) among hospitalized patients in Beijing (435).
This gene and variant were selected because of a prior candidate gene study by some of the same authors that found an association with influenza severity among Chinese patients during the 2009 influenza A H1N1/09 pandemic (436).
Here, they evaluated a small number (n=80) hospitalized COVID-19 patients to determine whether homozygosity for the previously identified risk allele was associated with mild versus severe disease (435).
They stated that they found an association between homozygosity for the SNP of interest and the severity of COVID-19.
A follow-up study demonstrated worldwide variation in the frequency of these SNPs (437), and subsequent studies claimed to support this result by comparing the frequency of the SNP in different groups to the COVID-19 case fatality rate in those groups; they examined SNPs in several candidate genes and identified an association with another SNP in IFITM3(438).
However, in the original study, the population-level frequency of the risk allele was consistent with its frequency in the mild population (436).
A similar analysis examined both SNPs in Britons of different ancestral backgrounds and also reported a correlation (439).
While this gene has been investigated for functions potentially relevant to COVID-19 pathogenesis by other groups as well (e.g., (440, 441), a follow-up analysis in Germany evaluated the effect of in 239 cases and 252 controls and reported non-significant effects (439).
The narrative surrounding IFITM3 therefore reflects a broad methodological critique about candidate gene studies, where results often fail to replicate (442).
The region associated with this gene was not identified in the large-scale GWAS conducted by the COVID-19 Host Genetics Initiative (COVID-19 HGI) (443), which is described in more detail below.
A second source of genetic variability that was hypothesized to have an effect on COVID-19 outcomes were human leukocyte antigens (HLA), or the major histocompatibility complex (MHC).
Both MHC classes I and II play a critical role in both the innate and adaptive immune system because they are a pivotal component of antigen presentation.
HLA classes I and II are also the most polymorphic loci in the human genome (444).
Additionally, because HLA polymorphisms are associated with geographic ancestry, study location and participant background offers important context (445).
Given the important role of the HLA complex in the immune response and the standing variation in the human population, HLA variation has been investigated for potential associations with COVID-19 outcomes.
Several approaches have been taken to evaluate a potential role of HLA in COVID-19.
In silico analysis suggested one particular HLA locus that could affect binding of SARS-CoV-2 peptides to MHC class II (446).
Other studies evaluated outcomes using retrospective cohort analyses.
An analysis of 95 South Asian COVID-19 patients found that HLA genotype was not significant in differentiating case severity when the necessary statistical corrections were applied (447).
Another study in a European population (n =147) did identify HLA alleles associated with severity (448).
In St. Louis, MO (USA), another study enrolled 234 COVID-19 cases, who were genotyped for HLA alleles and compared to a control population of 20,000 individuals from the National Marrow Donor Program (449).
They compared cases and controls on the basis of four “race/ethnic” populations and reported alleles showing a statistical association within each group (449).
However, because of this stratification, two of the demographic categories had less than ten cases.
Across all of these studies, there was minimal overlap in the risk alleles identified, and the small sample sizes raise concerns about the possibility for spurious hits.
The hypervariability of this region means that statistical power will necessarily be reduced, with much higher recruitment needed than for studies of biallelic loci.
A much larger analysis of 72,912 Israelis, 8.8% of whom tested positive for COVID-19, found no association between HLA genotype and infection or hospitalization (450).
Therefore, while MHC is functionally important to the immune response to COVID-19, it is not clear whether HLA genotypes are predictive of COVID-19 severity, and certainly such studies face exacerbated versions of the typical challenges of candidate gene studies.
Because of the challenges associated with analyzing such a variable region, it was excluded from the large-scale COVID-19 HGI GWAS analysis (443).
Finally, significant attention has been paid to the question of whether ABO blood type is associated with COVID-19 outcomes.
ABO blood type has been found to modulate susceptibility to other pathogens (451).
While ABO blood type is a genetic trait, it is more easily evaluated than the genetic regions discussed above because of the simple relationship between genetic variants and phenotype.
The possibility for an association between blood type and COVID-19 infection was raised early in the pandemic in a preprint that reported associations in 2,173 patients in Wuhan and Shenzhen, China (452).
The protective effect of O and increased risk associated with A blood types that they reported was subsequently investigated by many studies that returned varied results (e.g., (453–456); see (457) for a literature review).
Observations of higher and lower risk, respectively, of SARS-CoV-2 infection with A and O blood types was supported by a meta-analysis (458).
While the support for the association was independent of a mechanism, a possible relationship between ACE activity and blood type has been proposed (459) as has an effect on carbohydrate-carbohydrate interactions relevant to ACE2 binding (460).
This is the only candidate gene described that has received additional support from GWAS, as is discussed below.
The COVID-19 literature related to candidate gene investigations demonstrates relatively low inter-study consistency in findings.
In particular, sample size is a major challenge in designing these studies.
However, for many traits, the relationships between genes and phenotypes are complex, and selecting which variants to sequence is not always straightforward.
As a result, in the age of next-generation sequencing, discovery-driven studies have emerged as an alternative approach.
2.7.2 Genome-Wide Association Studies
Genome-wide association studies (GWAS) offers a discovery-driven approach that provides a different perspective than candidate gene studies.
Instead of selecting a gene or variant a priori, in GWAS, a large number of SNPs (usually several million) are evaluated at once to identify those most likely to vary in correlation with a trait of interest.
Because of the large number of statistical tests, statistical power and multiple hypothesis testing are both very important considerations in executing GWAS, which have also struggled with issues related to replicability (461).
In cases such as COVID-19 where outcomes can differ among ancestry groups (likely for non-genetic reasons, as reviewed in (339)), it is especially important that GWAS samples be selected with attention paid to ancestry, as incorrect or misleading associations can otherwise be identified with neutral markers indicative of ancestry itself (462).
Over the past two years, many GWAS have been undertaken with the aim of identifying variants associated with COVID-19 outcomes.
In some cases, the results have been consistent with hypothesized genetic correlates of susceptibility to COVID-19.
One study conducted a GWAS on a total of 435 COVID-19 patients from four countries and identified another HLA allele to be associated with an increased risk of intubation (463).
Other GWAS have identified an association with the ABO blood group locus.
One conducted a case/control GWAS in two populations, Italians and Spaniards, with 1980 cases and 2205 controls.
They reported two loci that met the genome-wide significance threshold, one on chromosome 3 and one on chromosome 9 (464).
The hit on chromosome 9 fell on the ABO locus and the alleles identified suggested a protective association with blood group O and a risk association with blood group A (464).
As the pandemic has progressed, large-scale efforts have been assembled to conduct GWAS on massive scales.
In March 2020, COVID-19 HGI was established as a world-wide consortium that combines data to conduct meta-analyses (465).
One year later, COVID-19 HGI released a meta-analysis of data from 46 studies, comprising 49,562 cases and 1,770,206 controls (443).
They identified 13 loci, seven of which were significant at the genome-wide level when considering all data available, that were associated with one or more phenotypes related to COVID-19 infection or severity.
Notably, strong signals were identified for both of the loci suggested by previous medium-scale GWAS in association with COVID-19 infection (464).
Additionally, several other loci could be mapped onto hypotheses about genetic contributors to immune function, lung function and disease.
This world-wide GWAS study made an effort towards strategic incorporation of genetic information from different ancestral groups.
Interestingly, the risk variant on chromosome 3 is likely to be inherited from Neanderthal introgression, meaning it is likely to be more prevalent in certain populations, especially non-African populations (466, 467).
The potential functional relationship between this region of the genome and COVID-19 is unknown, but phenome-wide association study has suggested blood cell traits as a potential trait regulated by this region (468).
Identifying genetic variants associated with a complex disease is always complicated.
In COVID-19 studies, the results of candidate gene analyses have in general been difficult to replicate.
However, large-scale collaboration on GWAS has made it possible to detect at least two loci that do appear to replicate across studies and potentially even across ancestral backgrounds.
2.8 Question 5: How is it Changing?
Evolution in SARS-CoV-2 has also been observed over a short timescale.
After zoonotic transfer, SARS-CoV-2 continued evolving in the human population (210).
The SARS-CoV-2 mutation rate is moderate compared to other RNA viruses (212), which likely restricts the pace of evolution in SARS-CoV-2.
Nevertheless, genomic analyses have yielded statistical evidence of ongoing evolution.
Initially, two known variants of the spike protein emerged that differed by a single amino acid at position 614 (G614 and D614), and there is evidence that G614 had become more prevalent than D614 by June 2020 (222).
While there is a hypothesis that this genomic change increased the SARS-CoV-2 infectivity and virulence, this hypothesis has not yet been tested due to a lack of data (469).
Another study (212) identified 198 recurrent mutations in a dataset of 7,666 curated sequences, all of which defined non-synonymous protein-level modifications.
This pattern of convergent evolution at some sites could indicate that certain mutations confer an adaptive advantage.
While it is evident that SARS-CoV-2 exhibits moderate potential for ongoing and future evolution, the relationship between mutations and pathogenicity is not yet known.
Additional data is needed in order to understand patterns of evolutionary change and the mechanisms by which they might affect virulence.
Several factors could promote the evolution of SARS-CoV-2, including host immunodeficiency and transient exposure to antibodies directed against SARS-CoV-2 proteins.
A single case study of SARS-CoV-2 infection in an immunocompromised female with chronic lymphocytic leukemia and hypogammaglobulinemia (470) suggested that an accelerated evolution of the virus could occur in conditions of immunodeficiency.
A first administration of convalescent plasma did not clear the virus, and an ensuing increase in the genomic diversity in the samples was observed, suggesting an accelerated evolution due to selection pressure.
A second administration of convalescent plasma cleared the virus from the host 105 days after the initial diagnosis.
However, throughout the duration of infection, the patient was asymptomatic but contagious.
A second single case study in a 45-year old male with antiphospholipid syndrome (471) confirmed the earlier results, providing evidence of persistent COVID-19 symptoms in an immunocompromised patient for 154 days following diagnosis, ultimately leading to the death of patient.
The treatments administered included remdesivir and the Regeneron anti-spike protein antibody cocktail.
Genomic analyses of the patient’s nasopharyngeal swabs confirmed an accelerated evolution of the virus through mutations in the spike gene and the receptor-binding domain.
In summary, these two case studies suggested an accelerated evolution and persistent shedding of the virus in conditions of immunodeficiency.
In particular, the first case highlighted the role of convalescent plasma in creating escape variants.
In fact, one study (472) exposed the SARS-CoV-2 virus to convalescent plasma in vitro repeatedly to see how much plasma was required to neutralize the virus.
The results of the first six exposures were similar, but they reported that after the seventh exposure (on day 45), the amount of plasma required began to increase.
In analyzing the viral variants present, they found that this viral escape was promoted by the sudden accumulation of mutations, especially in the receptor-binding domain (RBD) and N-terminal domain (NTD), that quickly rose in frequency.
By the thirteenth exposure (day 85), the virus had evolved three mutations and could no longer be neutralized by the plasma used, even though the plasma was comprised of polyclonal serum that targeted a variety of epitopes.
Taken together, these observations suggest that evolutionary analyses of SARS-CoV-2 can provide crucial information about the conditions that promote resistance in SARS-CoV-2 and the kinetics of how resistance develops, information which will be important for understanding the implications of how vaccine regimens are designed and whether/when next-generation vaccines will be needed.
When variants occur, they can rise in frequency by chance or through an adaptive process that confers a competitive advantage to the virus.
Variants that had the D614G mutation in the spike glycoprotein seemed to spread faster.
However, it has been suggested that the mutation rose in frequency due to early chance events rather than by adaptive events (473).
Another mutation, Y453F, that occurred in the receptor binding domain of S, was first detected in mink; however, the transmission to humans has been established.
In mink, this mutation conferred an advantage by increasing the affinity towards ACE2 (474).
Similarly, N501Y mutation induces an increased affinity towards human ACE2 and has been involved in the dominance of B.1.1.7 by outcompeting other variants (475).
Therefore, genomic surveillance is essential to prevent the emergence of super-spreaders (476).
Emerging methods are being applied to this problem in an effort to understand which mutations are most likely to be of significant concern.
Novel machine learning methods were developed to predict the mutations in the sequence that promote viral escape.
While they preserve the pathogenicity of the virus, escape mutations change the virus’s sequence to evade detection by the immune system.
By using tools from natural language processing (NLP), viral escape was modeled as an NLP problem (477) where a modification makes a sentence grammatically correct but semantically different.
Therefore, language models of viruses could predict mutations that change the presentation of the virus to the immune system but preserve its infectivity.
2.8.1 Variants of Concern and Variants under Surveillance
Viral replication naturally leads to the occurrence of mutations, and thus to genetic variation (478).
However, due to an intrinsic RNA proof-reading process in the SARS-CoV-2 virus, the pace of evolution of SARS-CoV-2 is moderate in comparison to other viruses (479).
The declaration of the first SARS-CoV-2 variant of concern (VOC) B.1.1.7 in December 2020 has attracted significant media attention.
While the B.1.1.7 lineage garnered attention in November 2020, two genomes of the lineage were detected as early as September 20th, 2020 from routine genomic data sampled in Kent (U.K.) by the COVID-19 Genomics UK Consortium (COG-UK).
The following day, a second B.1.1.7 genome was reported in greater London (234, 473, 480, 481)
Since then, B.1.1.7 has spread across the UK and internationally, and it has now been detected in at least 62 countries (482), despite several countries imposing travel restrictions on travelers from the UK.
Of the twenty-three mutations that define B.1.1.7 from the original strain isolated in Wuhan (lineage A), fourteen are lineage-specific and three appear to be biologically consequential mutations associated with the spike protein, namely N501Y, P681H, and 69-70del (480, 481).
The latter is a 6-bp deletion that leads to the loss of two amino acids and has consequences for immune recognition; it may, in conjunction with N501Y, be responsible for the increased transmissibility of the B.1.1.7 VOC due to changes in the RBD that increase binding affinity with ACE2 (230, 480).
B.1.1.7 has increased transmissibility by up to 56%, leading to an R0 of approximately 1.4.
Additionally, this VOC has been shown to be associated with increased disease severity and increased mortality (483).
Other variants also express the 69-70del mutation (484, 485), and public health officials in the United States and the UK have been able to use RT-PCR-based assays (ThermoFisher TaqPath COVID-19 assay) to identify sequences with this deletion because it occurs where the qPCR probe binds (234).
In the UK, B.1.1.7 is present in more than 97% of diagnostic tests that return negative for S-gene targets and positive for the other targets; thus, the frequency of S-gene target failure can be used as a proxy for the detection of B.1.1.7 (480, 486).
The FDA has highlighted that the performance of three diagnostic tests may be affected by the B.1.1.7 lineage because it could cause false negative tests (487).
While B.1.1.7 is currently the main VOC, other genetic variants also currently designated as VOCs have been detected, including B.1.351 and P.1, both of which emerged independently (488, 489).
B.1.351 was first detected in October 2020 in South Africa, was later detected in the EU on December 28th, 2020 and has now spread to at least 26 countries (231, 490, 491).
B.1.351 contains several mutations at the RBD including K417N, E484K, and N501Y.
While the biological significance of these mutations are still under investigation, it does appear that this lineage may be associated with increased transmissibility (492) due to the N501Y mutation (230, 481).
Additionally, an analysis of a pseudovirus expressing the 501Y.V2 spike protein (B.1.351) showed that this variant demonstrates increased resistance to neutralization by convalescent plasma, even though total binding activity remained mostly intact (493).
Further, using a live virus neutralization assay (LVNA), it was shown that 501Y.V2 (B.1.351) is poorly neutralized by convalescent plasma obtained from individuals who responded to non-501Y.V2 variants (494).
However, 501Y.V2 infection-elicited plasma was able to cross-neutralize earlier non-501Y.V2 variants, suggesting that vaccines targeting VOCs may be effective against other mutant lineages (494).
The P.1 variant is a sublineage of the B.1.1.28 lineage that was first detected in Japan in samples obtained from four travelers from Brazil during a screening at a Tokyo airport on January 10, 2021 (495).
Shortly thereafter, it was established that there was a concentration of cases of the P.1 variant in Manaus, Brazil.
In a small number of samples (n=31) sequenced in Manaus, 42% were identified as the P.1 variant as early as mid-December, but the variant seemed to be absent in genome surveillance testing prior to December (496).
To date, at least eight countries have detected the P.1 lineage (497).
While the majority of P.1 cases detected internationally have been linked to travel originating from Brazil, the UK has also reported evidence of community transmission detected via routine community sequencing (497, 498).
P.1 has eight lineage-specific mutations along with three concerning spike protein mutations in the RBD, including K417T, E484K, and N501Y (492).
There have been multiple different SARS-CoV-2 lineages detected that have mostly been of no more clinical concern than the original devastating lineage originating in Wuhan (499).
However, the spotlight has been cast on other variants of unknown clinical relevance due to the increase of cases observed that have been associated with B.1.1.7 in particular.
Although early in its ascendency, B.1.427/429 are SARS-CoV-2 variants that was detected in California, USA and also known as CAL.20C (500).
It was first detected in July 2020 but was not detected again until October 2020.
In December 2020, B.1.427/429 accounted for ~24% of the total cases in Southern California and ~36% of total cases in the Los Angeles area.
B.1.427/429 have now been detected in several U.S. states and at least 38 countries worldwide (500, 501).
This variant is characterized by five key lineage-specific mutations (ORF1a: I4205V, ORF1b:D1183Y, S: S13I;W152C;L452R).
The latter spike mutation, L452R, is found in an area of the RBD known to resist monoclonal antibodies to the spike protein (502), and it is hypothesized that this mutation may resist polyclonal sera in convalescent patients or in individuals post-vaccination (500, 503).
B.1.427/429 are now designated VOCs (489); however, further research is still required to determine the implications of the mutations encoded in this genetic variant.
Another notable variant has recently been discovered in 35 patients in a Bavarian hospital in Germany; however, the sequencing data has not been published to date and it remains to be determined whether this variant is of any further concern (504).
There are several shared mutations and deletions between the three lineages, P.1, B.1.1.7, and B.1.315 and indeed other variants of SARS-CoV-2 that are under investigation (496).
For example, N501Y, which appears to have occurred independently in each of the three lineages.
E484K is present in both B.1.351 and P.1 (505).
The mutations N501Y and E484K are found in the RBD within the receptor-binding motif responsible for forming an interface with the ACE2 receptor, which seems to be consequential for ACE2 binding affinity (506).
Indeed, N501Y is associated with increased virulence and infectivity in mouse models (507).
E484K has also been associated with evasion from neutralizing antibodies (472, 503, 508).
The del69-70 (del:11288:9) is also shared between P.1 and B.1.1.7 and happens to be a common deletion found in the N terminal mutation of the spike protein.
This deletion has also been associated with several RBD mutations (230, 481, 509).
There is concern that mutations in the spike protein of variants may lead to clinical consequences for transmissibility, disease severity, re-infection, therapeutics, and vaccinations (472, 503, 510–514).
Vaccine producers are working to determine whether the vaccines are still effective against the novel genetic variants.
Moderna recently published data for their mRNA-1273 vaccine that showed no significant impact of neutralization against the B.1.1.7 variant upon vaccination in humans and non-human primates.
On the other hand, Moderna reported a reduced but significant neutralization against the B.1.351 variant upon vaccination (515).
Indeed, Pfizer–BioNTech reported that sera from twenty participants vaccinated with the BNT162b COVID-19 vaccine in previous clinical trials (516, 517) elicited equivalent neutralizing titers against isogenic Y501 SARS-CoV-2 on an N501Y genetic background in vitro(518).
Another study has reported that the plasma neutralizing activity against SARS-CoV-2 variants encoding the combination of K417N:E484K:N501Y or E484K or N501Y was variably and significantly reduced in the sera of twenty participants who received either the Pfizer–BioNTech BNT162b (n = 6) vaccine or the Moderna’s mRNA-1273 vaccine (n =14) (519).
In a study focusing on serum samples from a combination of convalescent individuals, those who obtained the mRNA-1273 vaccine, and those who obtained Novavax, in comparison to the D614G variant, the B.1.419 variant was 2-3 times less sensitive to neutralization while the B.1.351 variant was 9-14 times less sensitive (520).
Indeed, the E484K substitution seen in the P.1 and B.1.315 variants of the B.1.1.7 lineage are broadly reported to substantially reduce the efficacy of mRNA-based vaccines (520–522).
For now, the consensus appears to be that the FDA-approved vaccines still seem to be generally effective against the genetic variants of SARS-CoV-2 and their accompanying mutations, albeit with a lower neutralizing capacity (515, 518, 519, 523), though select VOCs may present challenges.
Further research is required to discern the clinical, prophylactic, and therapeutic consequences of these genetic SARS-CoV-2 variants as the pandemic evolves.
2.9 SARS-CoV-2 Evolution and Vaccine Efficacy
With these vaccines in place, one concern is how the virus’s continued evolution will affect their efficacy.
Since the start of this pandemic, we have already seen multiple variants emerge: B.1.1.7, which emerged in the UK, B.1.351, which emerged in South Africa, and P.1, which emerged in Brazil.
Viruses evolve or mutate at different rates.
Mutation rate is measured as the number of substitutions per nucleotide per cell infected (μs/n/c) (524).
RNA viruses tend to have mutation rates between 10-6 to 10-4(524).
As a reference, influenza A virus has a mutation rate of 10-5, whereas the mutation rate of SARS-CoV-2 is lower, with the mutation rate estimated at 10-6(525).
The accumulation of mutations allows the virus to escape recognition by the immune system (526).
The efficacy of vaccines depends on their ability to train the immune system to recognize the virus.
Therefore, viruses can develop resistance to vaccines through the accumulation of mutations that affect recognition.
The lower mutation rate of SARS-CoV-2 suggests the possibility of SARS-CoV-2 vaccines having a more long-lasting effect compared to vaccines targeting the influenza A virus.
2.9.1 Alpha and Beta Variants
The current SARS-CoV-2 vaccines in distribution have been reported to provide similar efficacy against the B.1.1.7 variant compared to the variants common at the time they were developed but reduced efficacy against the B.1.351 variant (527).
Pfizer and Moderna announced that they are working on developing a booster shot to improve efficacy against the B.1.351 variant (528).
The WHO continues to monitor the emergence of variants and their impact on vaccine efficacy (529).
Previous research in the computational prediction of the efficacy of vaccines targeting the influenza A virus might complement efforts to monitor these types of viral outbreaks (530).
To adapt, future vaccines may need to account for multiple variants and strains of SARS-CoV-2, and booster shots may be required (531).
2.9.2 Delta Variant and Ct
One preprint (334) analyzed a retrospective cohort of patients in Singapore who contracted COVID-19 from April to June of 2021.
This study focused on those who were confirmed or inferred to have been infected by the Delta variant of concern, and its aim was to analyze virological kinetics.
They identified 218 cases, 71 (33%) of whom were fully vaccinated with either the Pfizer/BioNTech or Moderna mRNA vaccines, 13 (6%) of whom had received only one dose or had received the second dose less than two weeks prior to infection, and four (2%) of whom had received a vaccine developed with another technology.
Unvaccinated patients were more likely to be symptomatic or to progress to severe COVID-19 and showed more symptoms than vaccinated patients, despite the higher age of the vaccinated cohort.
Ct was assessed over disease course, although the specific procedures for when additional RT-PCR was conducted is not clear; however, it is stated that the data was smoothed based on day of illness.
There was no significant difference in median Ct in the initial samples taken from fully vaccinated and unvaccinated patients, but Ct increased (signifying reduced viral load) more rapidly in fully vaccinated patients.
Like most analyses analyzing Ct(1), this study does not provide the data to make conclusions about contagiousness, as the samples were not cultured.
All the same, these findings do suggest that vaccinated individuals may be able to clear the infection more quickly.
A second analysis was based in Dane County in Wisconsin, USA during summer 2021, when the Delta variant was known to be the dominant variant in the region (335).
According to Our World in Data (532), at the beginning of the study, 49.3% of residents of Dane County were fully vaccinated, with this number rising to 51.4% by the end of the study, although an earlier version of the preprint reported the vaccination rate in Dane County as 67.4%.
The authors identified no significant differences in Ct among fully vaccinated and unvaccinated cases.
The Ct thresholds reported were consistent with contagiousness as evaluated in other studies, and in the present study, SARS-CoV-2 could be cultured from 51 of 55 samples with Ct less than 25.
This study was not longitudinal, but the timing of testing relative to symptom onset between symptomatic vaccinated and unvaccinated patients.
The findings of this study are therefore consistent with the idea that vaccinated people are less likely to contract symptomatic or severe COVID-19, but in cases of breakthrough infection, are still likely to be able to transmit SARS-CoV-2 to others.
2.10 Question 6: What is Next?
The SARS-CoV-2 pandemic has presented many unprecedented scientific opportunities.
The rapid identification of the genomic sequence of the virus allowed for early contextualization of SARS-CoV-2 among other known respiratory viruses, and the scientific community has continued to collect, analyze, and disseminate information about the SARS-CoV-2 virus and the associated illness, COVID-19 at previously unimaginable rates (4).
The accessibility of genome sequencing technology has allowed for deep sequencing of the virus to establish a level of viral surveillance that had never before been achieved (214, 533, 534).
The information obtained from genetic, bioinformatics, and evolutionary analysis has played a significant role in shaping the global pandemic response (533, 535, 536).
For example, wastewater surveillance has emerged as a potential epidemiological tool to monitor SARS-CoV-2 spread over large regions, complementing clinical surveillance (537–539).
Humans shed SARS-CoV-2 viral RNA in feces (540) that can be detected in wastewater.
Protocols have been developed to safely and reproducibly isolate and quantify SARS-CoV-2 in samples obtained from wastewater processing plants (539, 541).
To date, studies show that wastewater surveillance is an effective tool to monitor SARS-CoV-2 spread over large sewersheds (537–539, 542).
Indeed, data from a study in New York City indicated that wastewater SARS-CoV-2 detection correlated with clinical detection of infection (542).
Similar studies have been conducted in Nevada (543) and Boston (544).
To date, studies have shown that factors such as temperature, the travel time of wastewater, and diurnal variability may affect detection of SARS-CoV-2 (537, 543).
Additionally, wastewater surveillance provides a tool to monitor fluctuations in the viral strains present in a community (545, 546).
Due to its demonstrated utility so far, the United States CDC established the National Wastewater Surveillance System (NWSS), which has emerged as an important surveillance tool for SARS-CoV-2 spread (547).
Knowledge of the evolution of SARS-CoV-2 is imperative to managing it moving forward (533, 548).
The evolutionary questions highlighted here all point back to the fact that efforts to prevent future epidemics and pandemics will benefit greatly from long-term, sustainable efforts to monitor disease.
Beyond understanding the status and evolution of known pathogens via genomic surveillance, greater preparedness for novel viral threats would also result from monitoring zoonotic disease.
If not addressed, economic and environmental stressors are likely to cause future zoonotic transfer of diseases in the future (549).
The COVID-19 pandemic has highlighted both the incredible insights available with modern evolutionary and genomic methodologies, but has also revealed the reluctance of political actors to commit resources to these efforts outside of periods of acute need.
The One Health framework has emerged from collaborations by many prominent non-governmental organizations such as the World Heath Organization to promote scientific goals supportive of pandemic preparedness (534).
Genomic surveillance of human pathogens and of pathogens at the human-wildlife interface is an important component needed to meet the goals of One Health (534).
These efforts are especially important as anthropogenic alterations to the landscape such as climate change and urbanization increase the risk of zoonotic disease transmission (550, 551).
With the COVID-19 pandemic serving as a clear illustration of why this surveillance is imperative and of its feasibility, wider awareness and adoption of the One Health paradigm is the last piece needed to develop practices that will prevent the next pandemic.
3 Molecular and Serologic Diagnostic Technologies for SARS-CoV-2
3.1 Abstract
The COVID-19 pandemic has presented many challenges that have spurred biotechnological research to address specific problems.
Diagnostics is one area where biotechnology has been critical.
Diagnostic tests play a vital role in managing a viral threat by facilitating the detection of infected and/or recovered individuals.
From the perspective of what information is provided, these tests fall into two major categories, molecular and serological.
Molecular diagnostic techniques assay whether a virus is present in a biological sample, thus making it possible to identify individuals who are currently infected.
Additionally, when the immune system is exposed to a virus, it responds by producing antibodies specific to the virus.
Serological tests make it possible to identify individuals who have mounted an immune response to a virus of interest and therefore facilitate the identification of individuals who have previously encountered the virus.
These two categories of tests provide different perspectives valuable to understanding the spread of SARS-CoV-2.
Within these categories, different biotechnological approaches offer specific advantages and disadvantages.
Here we review the categories of tests developed for the detection of the SARS-CoV-2 virus or antibodies against SARS-CoV-2 and discuss the role of diagnostics in the COVID-19 pandemic.
3.2 Importance
Testing is critical to pandemic management.
Among molecular tests, messaging about testing strategies has varied widely between countries, with the United States in particular emphasizing the higher sensitivity of polymerase chain reaction tests above immunoassays.
However, these tests offer different advantages, and a holistic view of the testing landscape is needed to identify the information provided by each test and its relevance to addressing different questions.
Another important consideration is the ease of use and ability to scale for each test, which determines how widely they can be deployed.
Here we describe the different diagnostic technologies available as well as the information they provide about SARS-CoV-2 and COVID-19.
3.3 Introduction
Since the emergence of Severe acute respiratory syndrome-like coronavirus 2 (SARS-CoV-2) in late 2019, significant international efforts have focused on managing the spread of the virus.
Identifying individuals who have contracted coronavirus disease 2019 (COVID-19) and may be contagious is crucial to reducing the spread of the virus.
Given the high transmissibility of SARS-CoV-2 and the potential for asymptomatic or presymptomatic individuals to be contagious (1), the development of rapid, reliable, and affordable methods to detect SARS-CoV-2 infection is and was vitally important for understanding and controlling spread.
For instance, test-trace-isolate procedures were an early cornerstone of many nations’ efforts to control the outbreak (552–554).
Such efforts depend on diagnostic testing.
The genetic sequence of the SARS-CoV-2 virus was first released by Chinese officials on January 10, 2020 (555), and the first test to detect the virus was released about 13 days later (556).
The genomic information was critical to the development of diagnostic approaches.
There are two main classes of diagnostic tests: molecular tests, which can diagnose an active infection by identifying the presence of SARS-CoV-2, and serological tests, which can assess whether an individual was infected in the past via the presence or absence of antibodies against SARS-CoV-2.
Over the course of the COVID-19 pandemic, a variety of tests have emerged within these two categories.
Molecular tests detect either viral RNA or protein in a patient sample.
They are essential to identifying infected individuals, which can be important for determining courses of action related to treatment, quarantine, and contact tracing.
Tests for viral RNA are done by reverse transcription (RT) of viral RNA to DNA followed by DNA amplification, usually with polymerase chain reaction (PCR) (557).
Tests for viral proteins typically use an antibody pair for detection as implemented in techniques such as lateral flow tests (LFTs) and enzyme-linked immunosorbent assays (ELISAs) (558, 559).
Molecular tests require the viral genome sequence in order to develop DNA primers for viral RNA detection or to express a viral protein for use as an antigen in antibody production.
Serological tests, on the other hand, detect the presence of antibodies in blood plasma samples or other biological samples, providing insight into whether an individual has acquired immunity against SARS-CoV-2.
Assays that can detect the presence of antibodies in blood plasma samples include ELISA, lateral flow immunoassay, and chemiluminescence immunoassay (CLIA) (560).
To distinguish past infection from vaccination, serological tests detect antibodies that bind the nucleocapsid protein of the SARS-CoV-2 virus (561).
They are useful for collecting population-level information for epidemiological analysis, as they can be used to estimate the extent of the infection in a given area.
Thus, serological tests may be useful to address population-level questions, such as the percent of cases that manifest as severe versus mild and for guiding public health and economic decisions regarding resource allocation and counter-disease measures.
Molecular and serological tests therefore offer distinct, complementary perspectives on COVID-19 infections.
Some of the same technologies are useful to both strategies, and different technologies have been employed to varying extents throughout the world since the start of the COVID-19 pandemic.
Two of the primary metrics used to evaluate these tests are sensitivity and specificity.
Sensitivity refers to a test’s ability to correctly identify a true positive; for example, a test with 50% sensitivity would identify SARS-CoV-2 in only one of every two positive samples.
On the other hand, specificity refers to how well a test is able to identify a negative sample as negative.
This metric can be relevant both in terms of understanding the risk of false positives and in discussing whether a test is susceptible to identifying other coronaviruses.
Here, we review the different types of tests within each category that have been developed and provide perspective on their applications.
3.4 Molecular Tests to Identify SARS-CoV-2
Molecular tests are used to identify distinct genomic subsequences of a viral molecule in a sample or the presence of viral protein, and they thus can be used to diagnose an active viral infection.
An important first step is identifying which biospecimens are likely to contain the virus in infected individuals and then acquiring these samples from the patient(s) to be tested.
Common sampling sources for molecular tests include nasopharyngeal cavity samples, such as throat washes, throat swabs, and saliva (562), and stool samples (563).
Once a sample is acquired from a patient, molecular tests detect SARS-CoV-2 based on the presence of either viral nucleic acids or viral proteins.
3.4.1 PCR-Based Tests
When testing for RNA from viruses like SARS-CoV-2, the first step involves pre-processing in order to create complementary DNA (cDNA) from the RNA sample using RT.
The second step involves the amplification of a region of interest in the cDNA using successive cycles of heating and cooling.
Depending on the application, this amplification is achieved using variations of PCR.
Reverse transcription polymerase chain reaction (RT-PCR) tests determine whether a target is present by amplifying a region of interest of cDNA (564).
Some tests use the results of the PCR itself (e.g., a band on a gel) to determine whether the pathogen is present.
However, this approach has not been employed widely in diagnostic testing, and instead most PCR-based tests are quantitative.
3.4.1.1 Quantitative Real-Time PCR
In contrast to RT-PCR, quantitative, real-time PCR uses fluorescent dyes that bind to the amplified DNA, thereby allowing a real time assessment of the amplification procedure (564) (in this manuscript we refer to quantitative real-time PCR as qPCR, following the Minimum Information for Publication of Quantitative Real-Time PCR Experiments guidelines (565), and when combined with reverse transcriptase steps, as is required for the evaluation of RNA, it is known as RT-qPCR.)
The time resolution provided by qPCR and RT-qPCR is useful because the amount of fluorescence emitted by the sample is proportional to the amount of DNA amplified, and therefore the amount of virus present can be indirectly measured using the cycle threshold (Ct) determined by qPCR.
The first test developed and validated for the detection of SARS-CoV-2 used RT-qPCR to detect several regions of the viral genome: the ORF1b of the RNA-dependent RNA polymerase (RdRP), the envelope protein gene (E), and the nucleocapsid protein gene (N) (556).
The publication reporting this test was released on January 23, 2020, less than two weeks after the sequence of the virus was first reported (556).
In collaboration with several other labs in Europe and in China, the researchers confirmed the specificity of this test with respect to other coronaviruses against specimens from 297 patients infected with a broad range of respiratory agents.
Specifically, this test uses two probes against RdRP, one of which is specific to SARS-CoV-2 (556).
Importantly, this assay was not found to return false positive results.
In January 2020, Chinese researchers developed a test that used RT-qPCR to identify two gene regions of the viral genome, ORF1b and N(566).
This assay was tested on samples from two COVID-19 patients and a panel of positive and negative controls consisting of RNA extracted from several cultured viruses.
The assay uses the N gene to screen patients, while the ORF1b gene region is used to confirm the infection (566).
The test was designed to detect sequences conserved across sarbecoviruses, or viruses within the same subgenus as SARS-CoV-2.
Considering that Severe acute respiratory syndrome-related coronavirus 1 (SARS-CoV-1) and SARS-CoV-2 are the only sarbecoviruses currently known to infect humans, a positive test can be assumed to indicate that the patient is infected with SARS-CoV-2, although this test is not able to discriminate the genetics of viruses within the sarbecovirus clade.
The fact that the targets are so conserved offers the advantage of reduced concern about sensitivity in light of the evolution of SARS-CoV-2.
qPCR tests have played an important role in diagnostics during the COVID-19 pandemic.
For SARS-CoV-2, studies have typically considered a patient to be infectious if the Ct is below 33 or sometimes 35 (1, 567, 568).
A lower Ct corresponds to fewer qPCR cycles needed to reach a detectable level, indicating that higher amounts of virus were present in the initial reaction.
Interpretations of the Ct values obtained from these tests have raised some interesting questions related to viral load and contagiousness.
Lower Ct values correspond to a higher probability of a positive viral culture, but no threshold could discriminate all positive from all negative cultures (240).
Additionally, because of the variability introduced by sample collection and clinical components of testing, Ct is not a proxy for viral load (569).
Positive PCR results have also been reported for extended periods of time from symptom onset and/or the first positive PCR test (275), meaning that in some cases, a positive PCR may not indicate that someone is contagious (1).
In addition to the nuance required to interpret PCR results, there are also factors that influence their accuracy.
The specificity of these tests is very high (570), meaning that a positive RT-PCR result is very likely to indicate SARS-CoV-2 infection.
The weight given to these tests as an indicator of SARS-CoV-2 infection regardless of other clinical considerations is not typical (571).
In fact, while the analytical specificity of the assay is extremely high, the challenges of implementing testing can introduce variability that results in a lower clinical specificity (571).
Several factors may influence the sensitivity and specificity, with sample collection being a critically important factor in the reliability of RT-PCR results.
The most reliable results were found to come from nasopharyngeal swabs and from pooled nasal and throat swabs, with lower accuracies produced by saliva or by throat or nasal swabs alone (570, 572).
Differences in experimental parameters such as the use of primers more specific to SARS-CoV-2 has been found to improve sensitivity in these specimens (573).
Additionally, the impact of viral evolution on RT-PCR sensitivity is a concern (574, 575).
Using a panel that includes multiple targets can mitigate these effects (576).
Additionally, a test designed to incorporate genomic differences with SARS-CoV-1 was found to offer improved sensitivity and specificity (573).
Thus, while various factors can influence the exact parameters of testing accuracy, RT-PCR is known to have very high specificity and lower, but still high, sensitivity.
3.4.1.2 Digital PCR
Digital PCR (dPCR) is a new generation of PCR technologies offering an alternative to traditional qPCR (577).
In dPCR, a sample is partitioned into thousands of compartments, such as nanodroplets (droplet dPCR or ddPCR) or nanowells, and a PCR reaction takes place in each compartment.
This design allows for a digital read-out where each partition is either positive or negative for the nucleic acid sequence being tested for, allowing for absolute target quantification through Poisson statistics.
While dPCR equipment is not yet as common as that for qPCR, dPCR for DNA targets generally achieves higher sensitivity than other PCR technologies while maintaining high specificity, though sensitivity is slightly lower for RNA targets (578).
High sensitivity is particularly relevant for SARS-CoV-2 detection, since low viral load in clinical samples can lead to false negatives.
In one study, Suo et al. (579) performed a double-blind evaluation of ddPCR for SARS-CoV-2 detection.
They evaluated on 63 samples collected from suspected positive outpatients and 14 from supposed convalescent patients.
Of the 63 outpatients, only 21 (33%) were identified as positive for SARS-CoV-2 with qPCR.
However, ddPCR identified 49 (78%) as positive, 10 (16%) as negative, and 4 (6%) as suspected/borderline for SARS-CoV-2 infection.
While both qPCR and ddPCR were found to have very high specificity (100%), this analysis reported that the sensitivity was 40% with qPCR compared to 94% with ddPCR.
Analysis of serial dilutions of a linear DNA standard suggested that ddPCR was approximately 500 times more sensitive than qPCR (579).
Thus, this study suggests that ddPCR provides an extremely sensitive molecular test that is able to detect SARS-CoV-2 even at very low viral loads.
A second study (580) confirmed that RT-ddPCR is able to detect SARS-CoV-2 at a lower threshold for viral load relative to RT-PCR.
This study analyzed 196 samples, including 103 samples from suspected patients, 77 from contacts and close contacts, and 16 from suspected convalescents, using both RT-qPCR and RT-ddPCR.
First, the authors evaluated samples from the 103 suspected cases.
Using RT-qPCR, 29 (28%) were identified as positive, 25 (24%) as negative, and 49 (48%) as borderline, i.e., the Ct value was higher than the positive threshold of 35 but lower than the negative threshold of 40.
When the 61 negative and borderline samples were reanalyzed with ddPCR, 19 (31%) of the negative and 42 (69%) of the borderline samples were identified as positive.
All of the suspected cases were later confirmed to be COVID-19 through a combination of symptom development and RT-qPCR resampling, indicating that ddPCR improved the overall detection rate compared to RT-qPCR from 28.2% to 87.4%.
They repeated this analysis in patient samples from contacts and close contacts.
Patients who tested negative with both methods (n = 48) were observed to remain healthy over a period of 14 days.
Among the remaining 29 samples from contacts, RT-qPCR identified 12 as positive, 1 as negative, and 16 as borderline.
All of the samples that tested positive using RT-qPCR also tested positive using ddPCR.
In contrast, the negative result and all but one of the borderline results were identified as positive by RT-ddPCR, and these patients were later determined to be SARS-CoV-2 positive based on clinical evaluation and repeated molecular sampling.
Similarly, in the final group, 16 convalescent patients, RT-qPCR identified 12 as positive, three as suspect, and one as negative, but RT-dPCR identified all as positive.
The evidence from this study therefore supports a lower limit of detection with ddPCR.
Overall, these studies suggest that ddPCR is a promising tool for overcoming the problem of false negatives in SARS-CoV-2 RNA testing, but this method is unlikely to affect the current pandemic due to its lack of availability.
3.4.1.3 Sequencing
In some cases, the DNA amplified with PCR is sequenced.
Sequencing requires an additional sample pre-processing step called library preparation.
Library preparation is the process of preparing the sample for sequencing, typically by fragmenting the sequences and adding adapters (581).
In some cases, library preparation can involve other modifications of the sample, such as adding barcodes to identify a particular sample within the sequence data.
Barcoding can therefore be used to pool samples from multiple sources.
There are different reagents used for library preparation that are specific to identifying one or more target sections with PCR (582).
Sequential pattern matching is then used to identify unique subsequences of the virus, and if sufficient subsequences are found, the test is considered positive.
Therefore, tests that use sequencing require a number of additional molecular and analytical steps relative to tests that use PCR alone.
Sequencing has been an important strategy for discovery of SARS-CoV-2 variants (e.g., see (500)).
Sequencing elucidates any genetic variants located between the PCR primers.
For this reason, it is critical to genomic surveillance efforts.
Genomic surveillance is an important complement to epidemiological surveillance efforts (583), as described below.
Through genomic surveillance, it has become possible to monitor the emergence of variants of interest and variants of concern (VOC) that may pose additional threats due to increased contagiousness, virulence, or immune escape (583, 584).
Sequencing also allows for analysis of the dominant strains in an area at a given time.
Worldwide, the extent of genomic surveillance varies widely, with higher-income countries typically able to sequence a higher percentage of cases (585).
Sequencing efforts are important for identifying variants containing mutations that might affect the reliability of molecular diagnostic tests, as well as mitigation measures such as therapeutics and prophylactics (574, 575).
Therefore, sequencing is an important component of diagnostics: while it is not necessary for diagnosing an individual case, it is critical to monitoring trends in the variants affecting a population and to staying aware of emerging variants that may pose additional challenges.
3.4.1.4 Pooled and Automated PCR Testing
Due to limited supplies and the need for more tests, several labs have found ways to pool or otherwise strategically design tests to increase throughput.
The first such result came from Yelin et al. (586), who reported that they could pool up to 32 samples in a single qPCR run.
This was followed by larger-scale pooling with slightly different methods (587).
Although these approaches are also PCR based, they allow for more rapid scaling and higher efficiency for testing than the initial PCR-based methods developed.
Conceptually, pooling could also be employed in analysis with RT-qPCR (588), and this strategy has been evaluated in settings such as schools (589) and hospitals (590).
3.4.2 RT-LAMP
RT-PCR remains the gold standard for detection of SARS-CoV-2 RNA from infected patients, but the traditional method requires special equipment and reagents, including a thermocycler.
Loop-mediated isothermal amplification (LAMP) is an alternative to PCR that does not require specialized equipment (591).
In this method, nucleic acids are amplified in a 25 μL reaction that is incubated and chilled on ice (591).
It uses primers designed to facilitate auto-cycling strand displacement DNA synthesis (591).
LAMP can be combined with reverse transcription (RT-LAMP) to enable the detection of RNA.
One study showed that RT-LAMP is effective for detection of SARS-CoV-2 with excellent specificity and sensitivity and that this method can be applied to unprocessed saliva samples (592).
This method was benchmarked against RT-PCR using 177 human nasopharyngeal RNA samples, of which 126 were COVID positive.
The authors break down the sensitivity of their test according to the Ct value from RT-PCR of the same samples; RT-LAMP performs at 100% sensitivity for samples with a Ct from RT-PCR of 32 or less.
The performance is worse when considering all RT-PCR positive samples (including those with Ct values between 32-40).
However, there is some evidence suggesting that samples obtained from individuals that achieve Ct values >30 measured using RT-PCR tend to be less infective that those that record a Ct value <30 (593–595), so RT-LAMP is still a useful diagnostic tool.
Various combinations of reagents are available, but one example is the WarmStart Colorimetric LAMP 2X Master Mix with a set of six primers developed previously by Zhang et al. (596).
To determine assay sensitivity, serial tenfold dilutions of in vitro transcribed N-gene RNA standard were tested using quantities from 105 copies down to 10 copies.
The assay readout is the color of the dye changing from pink to yellow due to binding to the DNA product over 30 minutes.
The RT-LAMP assay was then applied to clinical nasopharyngeal samples.
For viral loads above 100 copies of genomic RNA, the RT-LAMP assay had a sensitivity of 100% and a specificity of 96.1% from purified RNA.
The sensitivity of the direct assay of saliva by RT-LAMP was 85%.
Sensitivity and specificity metrics were obtained by comparison with results from RT-PCR.
RT-LAMP pilot studies for detection of SARS-CoV-2 were reviewed in a meta-analysis (597).
In the meta-analysis of all 2,112 samples, the cumulative sensitivity of RT-LAMP was calculated at 95.5%, and the cumulative specificity was 99.5%.
This test aims to bring the sensitivity of nucleic acid detection to the point of care or home testing setting.
It could be applied for screening, diagnostics, or as a definitive test for people who are positive based on LFTs (see below).
The estimated cost per test is about 2 euros when RNA extraction is included.
The main strength of this test over RT-PCR is that it can be done isothermally, but the main drawback is that it is about 10-fold less sensitive than RT-PCR.
The low cost, excellent sensitivity/specificity, and quick readout of RT-LAMP makes this an attractive alternative to RT-PCR.
Alternative strategies like RT-LAMP are needed to bring widespread testing away from the lab and into under-resourced areas.
3.4.3 CRISPR-based Detection
Technology based on CRISPR (clustered regularly interspaced short palindromic repeats) (598) has also been instrumental in scaling up testing protocols.
Two CRISPR-associated nucleases, Cas12 and Cas13, have been used for nucleic acid detection.
Multiple assays exploiting these nucleases have emerged as potential diagnostic tools for the rapid detection of SARS-CoV-2 genetic material and therefore SARS-CoV-2 infection.
The SHERLOCK method (Specific High-sensitivity Enzymatic Reporter unLOCKing) from Sherlock Biosciences relies on Cas13a to discriminate between inputs that differ by a single nucleotide at very low concentrations (599).
The target RNA is amplified by real-time recombinase polymerase amplification (RT-RPA) and T7 transcription, and the amplified product activates Cas13a.
The nuclease then cleaves a reporter RNA, which liberates a fluorescent dye from a quencher.
Several groups have used the SHERLOCK method to detect SARS-CoV-2 viral RNA.
An early study reported that the method could detect 7.5 copies of viral RNA in all 10 replicates, 2.5 copies in 6 out of 10, and 1.25 copies in 2 out of 10 runs (600).
It also reported 100% specificity and sensitivity on 114 RNA samples from clinical respiratory samples (61 suspected cases, among which 52 were confirmed and nine were ruled out by metagenomic next-generation sequencing, 17 SARS-CoV-2-negative but human coronavirus (HCoV)-positive cases, and 36 samples from healthy subjects) and a reaction turnaround time of 40 minutes.
A separate study screened four designs of SHERLOCK and extensively tested the best-performing assay.
They determined the limit of detection to be 10 copies/μl using both fluorescent and lateral flow detection (601).
LFT strips are simple to use and read, but there are limitations in terms of availability and cost per test.
Another group therefore proposed the CREST (Cas13-based, Rugged, Equitable, Scalable Testing) protocol, which uses a P51 cardboard fluorescence visualizer, powered by a 9-volt battery, for the detection of Cas13 activity instead of immunochromatography (602).
CREST can be run, from RNA sample to result, with no need for AC power or a dedicated facility, with minimal handling in approximately 2 hours.
Testing was performed on 14 nasopharyngeal swabs.
CREST picked up the same positives as the CDC-recommended TaqMan assay with the exception of one borderline sample that displayed low-quality RNA.
This approach may therefore represent a rapid, accurate, and affordable procedure for detecting SARS-CoV-2.
The DETECTR (DNA Endonuclease-Targeted CRISPR Trans Reporter) method from Mammoth Biosciences involves purification of RNA extracted from patient specimens, amplification of extracted RNAs by loop-mediated amplification, and application of their Cas12-based technology.
In this assay, guide RNAs (gRNAs) were designed to recognize portions of sequences corresponding to the SARS-CoV-2 genome, specifically the N2 and E regions (603).
In the presence of SARS-CoV-2 genetic material, sequence recognition by the gRNAs results in double-stranded DNA cleavage by Cas12, as well as cleavage of a single-stranded DNA molecular beacon.
The cleavage of this molecular beacon acts as a colorimetric reporter that is subsequently read out in a lateral flow assay and indicates the presence of SARS-CoV-2 genetic material and therefore SARS-CoV-2 infection.
The 40-minute assay is considered positive if there is detection of both the E and N genes or presumptive positive if there is detection of either of them.
The assay had 95% positive predictive agreement and 100% negative predictive agreement with the US Centers for Disease Control and Prevention SARS-CoV-2 RT-qPCR assay.
The estimated limit of detection was 10 copies per μl reaction, versus 1 copy per μl reaction for the CDC assay.
These results have been confirmed by other DETECTR approaches.
Using RT-RPA for amplification, another group detected 10 copies of synthetic SARS-CoV-2 RNA per μl of input within 60 minutes of RNA sample preparation in a proof-of-principle evaluation (604).
Through a similar approach, another group reported detection at 1 copy per μl (605).
The DETECTR protocol was improved by combining RT-RPA and CRISPR-based detection in a one-pot reaction that incubates at a single temperature and by using dual CRISPR RNAs, which increases sensitivity.
This new assay, known as All-In-One Dual CRISPR-Cas12a, detected 4.6 copies of SARS-CoV-2 RNA per μl of input in 40 minutes (606).
Another single-tube, constant-temperature approach using Cas12b instead of Cas12a achieved a detection limit of 5 copies/μl in 40-60 minutes (607).
It was also reported that electric field gradients can be used to control and accelerate CRISPR assays by co-focusing Cas12-gRNA, reporters, and target (608).
The authors generated an appropriate electric field gradient using a selective ionic focusing technique known as isotachophoresis (ITP) implemented on a microfluidic chip.
They also used ITP for automated purification of target RNA from raw nasopharyngeal swab samples.
Combining this ITP purification with loop-mediated isothermal amplification, their ITP-enhanced assay achieved detection of SARS-CoV-2 RNA (from raw sample to result) in 30 minutes.
All these methods require upstream nucleic acid amplification prior to CRISPR-based detection.
They rely on type V (Cas12-based) and type IV (Cas13-based) CRISPR systems.
In contrast, type III CRISPR systems have the unique property of initiating a signaling cascade, which could boost the sensitivity of direct RNA detection.
In type III CRISPR systems, guide CRISPR RNAs (crRNAs) are bound by several Cas proteins (609) and can target both DNA and RNA molecules (610, 611).
A study tested this hypothesis using the type III-A crRNA-guided surveillance complex from Thermus thermophilus(612).
The authors showed that activation of the Cas10 polymerase generates three products (cyclic nucleotides, protons, and pyrophosphates) that can all be used to detect SARS-CoV-2 RNA.
Detection of viral RNA in patient samples still required an initial nucleic acid amplification step, but improvements may in the future remove that requirement.
This goal of amplification-free detection was later achieved for a Cas13a-based system (613).
This approach combined multiple CRISPR RNAs to increase Cas13a activation, which is detected by a fluorescent reporter.
Importantly, because the viral RNA is detected directly, the test yields a quantitative measurement rather than a binary result.
The study also shows that fluorescence can be measured in a custom-made dark box with a mobile phone camera and a low-cost laser illumination and collection optics.
This approach is a truly portable assay for point-of-care diagnostics.
The authors achieved detection of 100 copies/μl of pre-isolated RNA in 30 minutes, and correctly identified all SARS-CoV-2-positive patient RNA samples tested in 5 minutes (n = 20).
There is an increasing body of evidence that CRISPR-based assays offer a practical solution for rapid, low-barrier testing in areas that are at greater risk of infection, such as airports and local community hospitals.
In the largest study to date, DETECTR was compared to RT-qPCR on 378 patient samples (614).
The authors reported 95% reproducibility.
Both techniques were equally sensitive in detecting SARS-CoV-2.
Lateral flow strips showed 100% correlation to the high-throughput DETECTR assay.
Importantly, DETECTR was 100% specific for SARS-CoV-2 and did not detect other human coronaviruses.
A method based on a Cas9 ortholog from Francisella novicida known as FnCas9 achieved 100% sensitivity and 97% specificity in clinical samples, and the diagnostic kit is reported to have completed regulatory validation in India (615).
3.4.4 Immunoassays for the Detection of Antigens
Immunoassays can detect molecular indicators of SARS-CoV-2 infection, such as the proteins that act as antigens from the SARS-CoV-2 virus.
They offer the advantage of generally being faster and requiring less specialized equipment than other molecular tests, especially those involving PCR.
As a result, immunoassays hold particular interest for implementation at home and in situations where resources for PCR testing are limited.
The trade-off is that these tests typically have a lower sensitivity, and sometimes a lower specificity, than other molecular tests.
However, these tests tend to return a positive result five to 12 days after symptom onset, which may therefore correlate more closely with the timeframe during which viral replication occurs (616).
Immunoassays for the detection of the SARS-CoV-2 antigen can include LFTs and ELISA, as discussed here, as well as CLIA and chromatographic immunoassays (617), as described in the serological testing section below.
3.4.4.1 Lateral Flow Tests
LFTs provide distinct value relative to PCR tests.
They can return results within 30 minutes and can be performed without specialized equipment and at low cost.
They also do not require training to operate and are cheap to produce.
Thus, they can be distributed widely to affected populations making them an important public health measure to curb pandemic spread.
LFTs rely on the detection of viral protein with an antibody.
Often this is done with an antibody sandwich format, where one antibody conjugated to a dye binds at one site on the antigen, and an immobilized antibody on the strip binds at another site (558).
This design allows the dye to accumulate to form a characteristic positive test line on the strip (558).
Outside of COVID-19 diagnostics, the applications of LFTs are broad; they are routinely used for home pregnancy tests, disease detection, and even drugs of abuse detection in urine (618).
A recent review surveyed the performance of LFTs for detection of current SARS-CoV-2 infection (619).
This review covered 24 studies that included more than 26,000 total LFTs.
They reported significant heterogeneity in test sensitivities, with estimates ranging from 37.7% to 99.2%.
The estimated specificities of these tests were more homogeneous, spanning 92.4% to 100.0%.
Despite having lower sensitivity than PCR tests, LFTs occupy an important niche in the management of SARS-CoV-2.
Current infection detection by LFTs enables the scale and speed of testing that is beneficial to managing viral spread.
LFTs were available freely to citizens in the United Kingdom until April 1, 2022 (620) and to citizens of the United States in early 2022 (621).
These tests are particularly useful for ruling out SARS-CoV-2 infection in cases where the likelihood of infection is low (e.g., asymptomatic individuals) and positives (including false positives) can be validated with testing by alternate means (622).
3.4.4.2 Enzyme-Linked Immunosorbent Assay
ELISA is a very sensitive immunoassay that can be considered a gold standard for the detection of biological targets, including antibodies and antigenic proteins (559).
It can be used to generate either quantitative or qualitative results that can be returned within a few hours (623).
ELISA builds on the idea that antibodies and antigens bind together to form complexes (559) and utilizes an enzyme covalently linked to an antibody against the antigen to produce assay signal, usually a color change (624).
The main advantage of ELISA is that it enables signal amplification through the enzyme’s activity, which increases sensitivity.
With sandwich ELISA, antibodies are immobilized on a surface such as a plate, and viral protein antigens in the sample bind and are retained (625).
A second antibody is added that binds to another site on the antigen is then added, and that second antibody is covalently linked to an enzyme.
A substrate for that enzyme is then added to produce signal, usually light or a color change
The exact strategy for tagging with a reporter enzyme varies among different types of ELISA (559, 625).
For COVID-19 diagnostics, ELISAs have been designed to detect the antigenic Spike protein (626).
One of these assays uses two monoclonal antibodies specific to the nucleocapsid of SARS-CoV-2 to evaluate the relationship between the effect of (estimated) viral load on the ability of the assay to detect the SARS-CoV-2 antigen (627).
This study analyzed 339 naso-oropharyngeal samples that were also analyzed with RT-qPCR as a gold standard.
RT-qPCR identified 147 samples as positive and 192 as negative.
The authors estimated the overall sensitivity and specificity to be 61.9% and 99.0%, respectively.
Sensitivity increased with higher Ct.
This study also assessed the performance of the ELISA test under different conditions in order to evaluate how robust it would be to the challenges of testing in real-world settings globally.
Higher sensitivity was achieved for samples that were stored under ideal conditions (immediate placement in -80° C).
Therefore, while immediate access to laboratory equipment is an advantage, it is not strictly necessary for ELISA to detect the antigen.
3.4.5 Limitations of Molecular Tests
Tests that identify SARS-CoV-2 using molecular technologies will identify only individuals with current infections and are not appropriate for identifying individuals who have recovered from a previous infection.
Among molecular tests, different technologies have different sensitivities and specificities.
In general, specificity is high, and even then, the public health repercussions of a false positive can generally be mitigated with follow-up testing.
On the other hand, a test’s sensitivity, which indicates the risk of a false-negative response, can pose significant challenge to large-scale testing.
False negatives are a significant concern for several reasons.
Importantly, clinical reports indicate that it is imperative to exercise caution when interpreting the results of molecular tests for SARS-CoV-2 because negative results do not necessarily mean a patient is virus-free (628).
To reduce occurrence of false negatives, correct execution of the analysis is crucial (629).
Additionally, PCR-based tests can remain positive for a much longer time than the virus is likely to be actively replicating (616), raising concerns about their informativeness after the acute phase of the disease.
Hence, the CDC has advised individuals who suspect they have been re-infected with SARS-CoV-2 to avoid using diagnostic tests within 90 days of receiving a previous positive test (630).
Additionally, the emerging nature of the COVID-19 pandemic has introduced some challenges related to uncertainty surrounding interactions between SARS-CoV-2 and its human hosts.
For example, viral shedding kinetics are still not well understood but are expected to introduce a significant effect of timing of sample collection on test results (629).
Similarly, the type of specimen could also influence outcomes, as success in viral detection varies among clinical sample types (570, 572, 629).
With CRISPR-based testing strategies, the gRNA can recognize off-target interspersed sequences in the viral genome (631), potentially resulting in false positives and a loss of specificity.
There are also significant practical and logistical concerns related to the widespread deployment of molecular tests.
Much of the technology used for molecular tests is expensive, and while it might be available in major hospitals and/or diagnostic centers, it is often not available to smaller facilities (632).
At times during the pandemic, the availability of supplies for testing, including swabs and testing media, has also been limited (633).
Similarly, processing times can be long, and tests might take up to 4 days to return results (632), especially during times of high demand, such as spikes in case numbers (634).
Countries have employed various and differing molecular testing strategies as a tool to reduce viral transmission, even among high-income countries (635).
The rapid development of molecular tests has provided a valuable, albeit imperfect, tool to identify active SARS-CoV-2 infections.
3.5 Serological Tests to Identify Recovered Individuals
Although several molecular diagnostic tests to detect viral genetic material have high specificity and sensitivity, they provide information only about active infection, and therefore offer just a snapshot-in-time perspective on the spread of a disease.
Most importantly, they would not work on a patient who has fully recovered from the virus at the time of sample collection.
In such contexts, serological tests are informative.
Serological tests use many of the same technologies as the immunoassays used to detect the presence of an antigen but are instead used to evaluate the presence of antibodies against SARS-CoV-2 in a serum sample.
These tests are particularly useful for insight into population-level dynamics and can also offer a glimpse into the development of antibodies by individual patients during the course of a disease.
Immunoassays can detect antibodies produced by the adaptive immune system in response to viral threat.
Understanding the acquisition and retention of antibodies is important both to the diagnosis of prior (inactive) infections and to the development of vaccines.
The two immunoglobulin classes that are most pertinent to these goals are immunoglobulin M (IgM), which are the first antibodies produced in response to an infection, and immunoglobulin G (IgG), which are the most abundant antibodies (636, 637).
Serological tests detect these antibodies, offering a mechanism through which prior infection can be identified.
However, the complexity of the human immune response means that there are many facets to such analyses.
In general, SARS-CoV-2 infection will induce the immune system to produce antibodies fairly quickly.
Prior research is available about the development of antibodies to SARS-CoV-1 during the course of the associated disease, severe acute respiratory syndrome (SARS).
IgM and IgG antibodies were detected in the second week following SARS-CoV-1 infection.
IgM titers peaked by the first month post-infection, and then declined to undetectable levels after day 180.
IgG titers peaked by day 60 and persisted in all donors through the two-year duration of study (638).
Such tests can also illuminate the progression of viral disease, as IgM are the first antibodies produced by the body and indicate that the infection is active.
Once the body has responded to the infection, IgG are produced and gradually replace IgM, indicating that the body has developed immunogenic memory (639).
Therefore, it was hoped that the development of assays to detect the presence of IgM and IgG antibodies against SARS-CoV-2 would allow the identification of cases from early in the infection course (via IgM) and for months or years afterwards (via IgG).
Several technologies have been used to develop serological tests for COVID-19, including ELISA, lateral flow immunoassay, chemiluminescence immunoassay, and neutralizing antibody assays (640).
3.5.1 ELISA
The application of ELISA to serological testing is complementary to its use in molecular diagnostics (see above).
Instead of using an enzyme-labeled antibody as a probe that binds to the target antigen, the probe is an antigen and the target is an antibody.
The enzyme used for detection and signal amplification is on a secondary antibody raised generally against human IgG or IgM.
In March 2020, the Krammer lab proposed an ELISA test that detects IgG and IgM that react against the receptor-binding domain (RBD) of the spike proteins (S) of the virus (641).
A subsequent ELISA test developed to detect SARS-CoV-2 IgG based on the RBD reported a specificity of over 99% and a sensitivity of up to 88.24%, which was observed in samples collected 21 to 27 days after the onset of infection (approximated with symptom onset or positive PCR test) (642).
Earlier in the disease course, sensitivity was lower: 53.33% between days 0 and 13 and 80.47% between days 14 and 20.
This study reported that their laboratory ELISA outperformed two commercial kits that also used an ELISA design (642).
Therefore, while analysis with ELISA requires laboratory support and equipment, these results do suggest that ELISA achieves relatively high sensitivity, especially in the weeks following infection.
Efforts have been made to develop low-cost strategies for conducting these tests that will make them more accessible worldwide (643).
3.5.2 Chemiluminescence Immunoassay
Another early approach investigated for detection of antibodies against SARS-CoV-2 was CLIA.
Like ELISA, CLIA is a type of enzyme immunoassay (EIA) (644).
While the technique varies somewhat, in one approach, a bead is coated with the antigen and then washed with the sample (645).
If the antibody is present in the sample, it will bind to the bead.
Then the bead is exposed to a label, a luminescent molecule that will bind to the antigen/antibody complex and can therefore be used as an indicator (645).
One CLIA approach to identify COVID-19 used a synthetic peptide derived from the amino acid sequence of the SARS-CoV-2 S protein (646).
It was highly specific to SARS-CoV-2 and detected IgM in 57.2% and IgG in 71.4% of serum samples from 276 COVID-19 cases confirmed with RT-qPCR.
IgG could be detected within two days of the onset of fever, but IgM could not be detected any earlier (646), which has been supported by other analyses as well (647).
This pattern was consistent with observations in Middle East respiratory syndrome, which is also caused by an HCoV.
In comparisons of different commercial immunoassays, accuracy of CLIA tests were often roughly comparable to other EIAs (648), although one CLIA did not perform as well as several other EIAs (647, 649).
The sensitivity and specificities reported vary among CLIA tests and for the detection of IgM versus IgG, but sensitivities and specificities as high as 100% have been reported among various high-throughput tests (649–651).
CLIA has previously been used to develop tests that can be used at point of care (e.g., (644)) which may allow for this technique to become more widely accessible in the future.
3.5.3 Lateral Flow Immunoassay
The first serological test approved for emergency use in the United States was developed by Cellex (652).
The Cellex qSARS-CoV-2 IgG/IgM Rapid Test is a chromatographic immunoassay, also known as a lateral flow immunoassay, designed to qualitatively detect IgM and IgG antibodies against SARS-CoV-2 in the plasma of patients suspected to have developed a SARS-CoV-2 infection (652).
The Cellex test cassette contains a pad of SARS-CoV-2 antigens and a nitrocellulose strip with lines for each of IgG and IgM, as well as a control (goat IgG) (652).
In a specimen that contains antibodies against the SARS-CoV-2 antigen, the antibodies will bind to the strip and be captured by the IgM and/or IgG line(s), resulting in a change of color (652).
With this particular assay, results can be read within 15 to 20 minutes (652).
Lateral flow immunoassays are often available at point of care but can have very low sensitivity (649).
3.5.4 Neutralizing Antibody Assays
Neutralizing antibody assays play a functional role in understanding immunity that distinguishes them from other serological tests.
The tests described above are all binding antibody tests.
On the other hand, rather than simply binding an antibody to facilitate detection, neutralizing antibody assays determine whether an antibody response is present that would prevent infection (653, 654).
Therefore, these tests serve the purpose of evaluating the extent to which a sample donor has acquired immunity that will reduce susceptibility to SARS-CoV-2.
As a result, neutralizing antibody assays have been used widely to characterize the duration of immunity following infection, to assess vaccine candidates, and to establish correlates of protection against infection and disease (655–657).
These tests are typically performed in a laboratory (653), and in SARS-CoV-2, the results of neutralizing antibody assays are often correlated with the results of binding antibody tests (653).
The gold standard for assessing the presence of neutralizing antibodies is the plaque reduction neutralization test (PRNT), but this approach does not scale well (654).
An early high-throughput neutralizing antibody assay designed against SARS-CoV-2 used a fluorescently labeled reporter virus that was incubated with different dilutions of patient serum (654).
The cells used for incubation would turn green if antibodies were not present.
Essentially, this assay evaluates whether the virus is able to infect the cell in the presence of the serum.
The specificity of this assay was 100%, and the correlation between the results of this assay and of PRNT was 0.85 with the results suggesting that the sensitivity of the high-throughput approach was higher than that of PRNT (654).
While this approach was performed on a plate and using cells, other methods have been developed using methods such as bead arrays (658).
3.5.5 Duration of Immune Indicators
While the adaptive immune system produces antibodies in response to SARS-CoV-2 viral challenge, these indicators of seroconversion are unlikely to remain in circulation permanently.
Previously, a two-year longitudinal study following convalesced SARS patients with a mean age of 29 found that IgG antibodies were detectable in all 56 patients surveyed for at least 16 months and remained detectable in all but 4 patients (11.8%) through the full two-year study period (659).
These results suggest that immunity to SARS-CoV-1 is sustained for at least a year.
Circulating antibody titers to other coronaviruses have been reported to decline significantly after one year (660).
Evidence to date suggests that sustained immunity to the SARS-CoV-2 virus remains for a shorter period of time but at least 6 to 8 months after infection (661–664).
However, this does not mean that all serological evidence of infection dissipates, but rather that the immune response becomes insufficient to neutralize the virus.
In order to study the persistence of SARS-CoV-2 antibodies, one study assessed sustained immunity using 254 blood samples from 188 COVID-19 positive patients (662).
The samples were collected at various time points between 6 and 240 days post-symptom onset; some patients were assessed longitudinally.
Of the samples, 43 were collected at least 6 months after symptom onset.
After one month, 98% of patients were seropositive for IgG to S.
Moreover, S IgG titers were stable and heterogeneous among patients over a period of 6 to 8 months post-symptom onset, with 90% of subjects seropositive at 6 months.
Similarly, at 6 to 8 months 88% of patients were seropositive for RBD IgG, and 90% were seropositive for SARS-CoV-2 neutralizing antibodies.
Another study examined 119 samples from 88 donors who had recovered from mild to severe cases of COVID-19 (664).
A relatively stable level of IgG and plasma neutralizing antibodies was identified up to 6 months post diagnosis.
Significantly lower but considerable levels of anti-SARS-CoV-2 IgG antibodies were still present in 80% of samples obtained 6 to 8 months post-symptom onset.
Titers of IgM and IgG antibodies against the RBD were found to decrease from 1.3 to 6.2 months post infection in a study of 87 individuals (665).
However, the decline of IgA activity (15%) was less pronounced than that of IgM (53%) or IgG (32%).
It was noted that higher levels of anti-RBD IgG and anti-N total antibodies were detected in individuals that reported persistent post-acute symptoms at both study visits.
Moreover, plasma neutralizing activity decreased five-fold between 1.3 and 6.2 months in an assay of HIV-1 virus pseudotyped with SARS-CoV-2 S protein, and this neutralizing activity was directly correlated with IgG anti-RBD titers (665).
These findings are in accordance with other studies that show that the majority of seroconverters have detectable, albeit decreasing, levels of neutralizing antibodies at least 3 to 6 months post infection (666–668).
Determining the potency of anti-RBD antibodies early in the course of an infection may be important moving forward, as their neutralizing potency may be prognostic for disease severity and survival (669).
The duration of immunity might also vary with age (670) or ABO blood type (671).
Autopsies of lymph nodes and spleens from severe acute COVID-19 patients showed a loss of T follicular helper cells and germinal centers that may explain some of the impaired development of antibody responses (672).
Therefore, serological testing may be time-limited in its ability to detect prior infection.
Other immune indicators of prior infection have also been evaluated to see how they persist over time.
SARS-CoV-2 memory CD8+ T cells were slightly decreased (50%) 6 months post-symptom onset.
In this same subset of COVID-19 patients, 93% of subjects had detectable levels of SARS-CoV-2 memory CD4+ T cells, of which 42% had more than 1% SARS-CoV-2-specific CD4+ T cells.
At 6 months, 92% of patients were positive for SARS-CoV-2 memory CD4+ T cells.
Indeed, the abundance of S-specific memory CD4+ T cells over time was similar to that of SARS-CoV-2-specific CD4+ T cells overall (662).
T cell immunity to SARS-CoV-2 at 6 to 8 months following symptom onset has also been confirmed by other studies (664, 673, 674).
In another study, T cell reactivity to SARS-CoV-2 epitopes was also detected in some individuals never been exposed to SARS-CoV-2.
This finding suggests the potential for cross-reactive T cell recognition between SARS-CoV-2 and pre-existing circulating HCoV that are responsible for the “common cold” (675), but further research is required.
Therefore, whether T cells will provide a more stable measure through which to assess prior infection remains unknown.
Notably, commercial entities have tried to develop tests specifically for T cells, some of which have been authorized by the United States Food and Drug Administration (676, 677) to identify people with adaptive T cell immune responses to SARS-CoV-2, either from a previous or ongoing infection.
3.5.6 Applications of Serological Tests
In addition to the limitations posed by the fact that antibodies are not permanent indicators of prior infection, serological immunoassays carry a number of limitations that influence their utility in different situations.
Importantly, false positives can occur due to cross-reactivity with other antibodies according to the clinical condition of the patient (652).
Due to the long incubation times and delayed immune responses of infected patients, serological immunoassays are insufficiently sensitive for a diagnosis in the early stages of an infection.
Therefore, such tests must be used in combination with RNA detection tests if intended for diagnostic purposes (678).
False positives are particularly harmful if they are erroneously interpreted to mean that a population is more likely to have acquired immunity to a disease (679).
Similarly, while serological tests may be of interest to individuals who wish to confirm they were infected with SARS-CoV-2 in the past, their potential for false positives means that they are not currently recommended for this use.
However, in the wake of vaccines becoming widely available, accurate serological tests that could be administered at point of care were investigated in the hope that they could help to prioritize vaccine recipients (680).
Another concern with serological testing is the potential for viral evolution to reduce the sensitivity of assays, especially for neutralizing antibody assays.
Chen et al. performed a systematic re-analysis of published data examining the neutralizing effect of serum from vaccinated or recovered individuals on four VOC (681).
They found reduced neutralizing titers against these variants relative to the lineages used for reference.
These findings suggest that such techniques will need to be modified over time as SARS-CoV-2 evolves.
These limitations make serological tests far less useful for diagnostics and for test-and-trace strategies; however, serological testing is valuable for public health monitoring at the population level.
Serosurveys provide a high-level perspective of the prevalence of a disease and can provide insight into the susceptibility of a population as well as variation in severity, e.g., between geographic regions (679).
From a public health perspective, they can also provide insight into the effectiveness of mitigation efforts and to gain insight into risk factors influencing susceptibility (682).
EIA methods are high-throughput (683, 684), and, as with molecular tests, additional efforts have been made to scale up the throughput of serological tests (685).
Therefore, serological tests can be useful to developing strategies for the management of viral spread.
Early in the course of the pandemic, it was also hoped that serological tests would provide information relevant to advancing economic recovery.
Some infectious agents can be controlled through “herd immunity”, which is when a critical mass within the population acquires immunity through vaccination and/or infection, preventing an infectious agent from spreading widely.
It was hoped that people who had recovered and developed antibodies might be able to return to work (686, 687).
This strategy would have relied on recovered individuals acquiring long-term immunity, which has not been borne out (688).
Additionally, it was hoped that identifying seroconverters and specifically those who had mounted a strong immune response would reveal strong candidates for convalescent plasma donation (641); however, convalescent plasma has not been found to offer therapeutic benefit (reviewed in (3)).
While these hopes have not been realized, serological tests have been useful for gaining a better understanding of the pandemic (682).
3.6 Possible Alternatives to Current Diagnostic Practices
One possible alternative or complement to molecular and serological testing would be diagnosing COVID-19 cases based on symptomatology.
COVID-19 can present with symptoms similar to other types of pneumonia, and symptoms can vary widely among COVID-19 patients; therefore, clinical presentation is often insufficient as a sole diagnostic criterion.
In addition, identifying and isolating mild or asymptomatic cases is critical to efforts to manage outbreaks.
Even among mildly symptomatic patients, a predictive model based on clinical symptoms had a sensitivity of only 56% and a specificity of 91% (689).
More problematic is that clinical symptom-based tests are only able to identify already symptomatic cases, not presymptomatic or asymptomatic cases.
They may still be important for clinical practice and for reducing tests needed for patients deemed unlikely to have COVID-19.
In some cases, clinical signs may also provide information that can inform diagnosis.
Using computed tomography of the chest in addition to RT-qPCR testing was found to provide a higher sensitivity than either measure alone (690).
X-ray diagnostics have been reported to have high sensitivity but low specificity in some studies (691).
Other studies have shown that specificity varies between radiologists (692), though the sensitivity reported here was lower than that published in the previous paper.
While preliminary machine-learning results suggested that chest X-rays might provide high sensitivity and specificity and potentially facilitate the detection of asymptomatic and presymptomatic infections (e.g., (693)), further investigation suggested that such approaches are prone to bias and are unlikely to be clinically useful (694).
Given the above, the widespread use of X-ray tests on otherwise healthy adults is likely inadvisable.
Finally, in addition to genomic and serological surveillance, other types of monitoring have proven useful in managing the pandemic (695).
One that has received significant attention is wastewater surveillance.
This approach can use several of the technologies described for molecular testing, such as qPCR and dPCR, as well as in vitro culturing (696) and can provide insight into trends in the prevalence of SARS-CoV-2 regionally.
3.7 Strategies and Considerations for Testing
Deciding whom to test, when to test, and which test to use have proven challenging as the COVID-19 pandemic has unfolded.
Early in the COVID-19 pandemic, testing was typically limited to individuals considered high risk for developing serious illness (697).
This approach often limited testing to people with severe symptoms and people showing mild symptoms that had been in contact with a person who had tested positive.
Individuals who were asymptomatic (i.e., potential spreaders) and individuals who were able to recover at home were thus often unaware of their status.
Therefore, this method of testing administration misses a high proportion of infections and does not allow for test-and-trace methods to be used.
For instance, a study from Imperial College estimates that in Italy, the true number of infections was around 5.9 million in a total population of ~60 million, compared to the 70,000 detected as of March 28, 2020 (314).
Another analysis, which examined New York state, indicated that as of May 2020, approximately 300,000 cases had been reported in a total population of approximately 20 million (698).
This corresponded to ~1.5% of the population, but ~12% of individuals sampled statewide were estimated as positive through antibody tests (along with indications of spatial heterogeneity at higher resolution) (698).
Technological advancements that facilitate widespread, rapid testing would therefore be valuable for accurately assessing the rate of infection and aid in controlling the virus’ spread.
Additionally, the trade off of accessibility, sensitivity, and time to results has raised some complex questions around which tests are best suited to certain situations.
Immunoassays, including serological tests, have much higher limits of detection than PCR tests do (699).
Changes in public attitudes and the lifting of COVID-19 restrictions due to the multifactorial desire to stimulate economic activities has required a shift of testing paradigms in 2022, despite warnings from public health officials against a hard exit from public health restrictions (700, 701).
An important strategy for testing moving forward is to determine when someone becomes infectious or is no longer infectious following a positive test for COVID-19.
Generally, patient specimens tend to not contain culturable virus past day 5 of symptom onset (702, 703).
However, due to their sensitivity to post-infectious viral RNA in specimens, PCR-based methods may mislead individuals to believe that they are still infectious several days after symptom onset (678).
Furthermore, detection of viral RNA can occur days and weeks after an active infection due to the sensitivity of PCR-based methods (568, 704, 705).
In contrast, LFTs were thought to have poor sensitivity and their value for identifying infections and managing the pandemic was questioned (706, 707).
However, LFTs do reliably detect SARS-CoV-2 proteins when there is a high viral load, which appears to correlate with a person’s infectiousness (616, 708).
Therefore, LFTs are an important diagnostic tool to determine infectiousness with fast turnaround times, ease of use, and accessibility by the general public (678, 709).
One study has suggested that the test sensitivity of LFTs appears to be less important than accessibility to LFTs, frequent testing, and fast reporting times for reducing the impact of viral spread (710).
While PCR-based methods are important for COVID-19 surveillance, their use is labor intensive and time consuming, and laboratories are often slow to report results, rendering such methods limited in their surveillance capacity (678).
These limitations are demonstrated by the estimated 10-fold under-reporting of cases in the United States in 2020 due to shortages in testing and slow rollout of testing and slow reporting of results (711).
However, one strategy that may balance the strengths and weaknesses of both types of tests is to corroborate a positive LFT result using a PCR-based method.
Indeed, in December 2021 sufficient surveillance and reduction of COVID-19 spread using this joint LFT-PCR strategy was demonstrated in Liverpool, U.K., where there was an estimated 21% reduction of cases (709, 712).
3.8 What Lies Ahead
Diagnostic tools have played an important role during the COVID-19 pandemic.
Different tests offer different advantages (Figure 2).
Specifically, the results of SARS-CoV-2 diagnostic tests (typically qPCR or LFT-based tests) have been used to estimate the number of infections in the general population, thus informing public health strategies around the globe (574).
During the surges caused by the different SARS-CoV-2 variants between 2020 and 2021, government-sponsored efforts to conduct mass testing and to provide free diagnostic tests to the population were a common occurrence in many parts of the world (713–715).
However, recent reports indicate that such public health policies are starting to change during 2022.
For example, it is known that the UK plans to dismantle its COVID-19 testing program and scale back its daily reporting requirements (716, 717).
A similar approach can be seen in the US as well, where multiple state-run testing facilities are closing, despite some groups advocating to keep them open (718, 719).
These ongoing changes in testing policy are likely to have a direct effect on how the pandemic is managed moving forward.
SARS-CoV-2 diagnostic tests can be used effectively to slow the spread of the disease only when 1) they are used to share testing results in a timely manner so that they can reasonably be used to approximate the number of infections in the population and 2) those tests are easily accessible by the general public.
Figure 2:Summary of Diagnostic Technologies used in COVID-19 Testing.
The immune response to SARS-CoV-2 means that different diagnostic approaches offer different views of COVID-19.
Early in the infection course, viral load is high.
This means that PCR-based testing and EIA testing for antigens are likely to return positives (as indicated by the green bars at the bottom).
As viral load decreases, EIA antigen tests become negative, but PCR-based tests can still detect even very low viral loads.
From a serological perspective, IgM peaks in the first few weeks following infection and then decreases, while IgG peaks much later in the infection course.
Therefore, serological tests are likely to return positives in first few months following the acute infection course.
Additional detail is available above and in several analyses and reviews (1, 678, 705, 720).
Children are one segment of the population where the importance of the two aforementioned conditions can be exemplified.
This group is particularly vulnerable as there are ongoing challenges with testing in schools, increased COVID-19 mortality rates, and COVID-19-associated orphanhood.
In this regard, although there is evidence of the efficacy of routine diagnostic testing to reduce the probability of having infectious students (721, 722), as of March of 2022 there is an increasing number of schools that have stopped or plan to stop contact tracing efforts (723, 724), in line with an announcement made by the CDC where it no longer recommended contact tracing as a strategy to contain the virus (725).
An estimated 197 children have died in the US from COVID-19 during the first three months of 2022 (726), compared to 735 deaths in the preceding 20 months of the pandemic (727), and millions of children have been orphaned as a consequence of parent or caregiver death due to COVID-19 (728).
It is likely that reducing or eliminating testing capacity in schools will directly exacerbate these negative outcomes for the remainder of 2022.
The SARS-CoV-2 diagnostic tools presented in this paper are far less useful if they are difficult to obtain, or if their limited use results in biased data that would lead to ill-informed public health strategies.
Under conditions of limited supply, different strategies for testing are needed (729).
The pandemic is still an ongoing public health threat and it is worrying that active testing and tracing efforts are a low priority for public health authorities in many countries.
If this trend continues, the lack of testing could result in increased morbidity and mortality and an overall failure to manage the pandemic.
4 Identification and Development of Therapeutics for COVID-19
4.1 Abstract
After emerging in China in late 2019, the novel coronavirus SARS-CoV-2 spread worldwide and as of mid-2021 remains a significant threat globally.
Only a few coronaviruses are known to infect humans, and only two cause infections similar in severity to SARS-CoV-2: Severe acute respiratory syndrome-related coronavirus, a closely related species of SARS-CoV-2 that emerged in 2002, and Middle East respiratory syndrome-related coronavirus, which emerged in 2012.
Unlike the current pandemic, previous epidemics were controlled rapidly through public health measures, but the body of research investigating severe acute respiratory syndrome and Middle East respiratory syndrome has proven valuable for identifying approaches to treating and preventing novel coronavirus disease 2019 (COVID-19).
Building on this research, the medical and scientific communities have responded rapidly to the COVID-19 crisis to identify many candidate therapeutics.
The approaches used to identify candidates fall into four main categories: adaptation of clinical approaches to diseases with related pathologies, adaptation based on virological properties, adaptation based on host response, and data-driven identification of candidates based on physical properties or on pharmacological compendia.
To date, a small number of therapeutics have already been authorized by regulatory agencies such as the Food and Drug Administration (FDA), while most remain under investigation.
The scale of the COVID-19 crisis offers a rare opportunity to collect data on the effects of candidate therapeutics.
This information provides insight not only into the management of coronavirus diseases, but also into the relative success of different approaches to identifying candidate therapeutics against an emerging disease.
4.2 Importance
The COVID-19 pandemic is a rapidly evolving crisis.
With the worldwide scientific community shifting focus onto the SARS-CoV-2 virus and COVID-19, a large number of possible pharmaceutical approaches for treatment and prevention have been proposed.
What was known about each of these potential interventions evolved rapidly throughout 2020 and 2021.
This fast-paced area of research provides important insight into how the ongoing pandemic can be managed and also demonstrates the power of interdisciplinary collaboration to rapidly understand a virus and match its characteristics with existing or novel pharmaceuticals.
As illustrated by the continued threat of viral epidemics during the current millennium, a rapid and strategic response to emerging viral threats can save lives.
In this review, we explore how different modes of identifying candidate therapeutics have borne out during COVID-19.
4.3 Introduction
The novel coronavirus Severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) emerged in late 2019 and quickly precipitated the worldwide spread of novel coronavirus disease 2019 (COVID-19).
COVID-19 is associated with symptoms ranging from mild or even asymptomatic to severe, and up to 2% of patients diagnosed with COVID-19 die from COVID-19-related complications such as acute respiratory disease syndrome (ARDS) (1).
As a result, public health efforts have been critical to mitigating the spread of the virus.
However, as of mid-2021, COVID-19 remains a significant worldwide concern (Figure 3), with 2021 cases in some regions surging far above the numbers reported during the initial outbreak in early 2020.
While a number of vaccines have been developed and approved in different countries starting in late 2020 (5), vaccination efforts have not proceeded at the same pace throughout the world and are not yet close to ending the pandemic.
Due to the continued threat of the virus and the severity of the disease, the identification and development of therapeutic interventions have emerged as significant international priorities.
Prior developments during other recent outbreaks of emerging diseases, especially those caused by human coronaviruses (HCoV), have guided biomedical research into the behavior and treatment of this novel coronavirus infection.
However, previous emerging HCoV-related disease threats were controlled much more quickly than SARS-CoV-2 through public health efforts (Figure 3).
The scale of the COVID-19 pandemic has made the repurposing and development of pharmaceuticals more urgent than in previous coronavirus epidemics.
4.3.1 Lessons from Prior HCoV Outbreaks
Figure 3:Cumulative global incidence of COVID-19 and SARS.
As of March 9, 2023, 676,570,149 COVID-19 cases and 6,881,802 COVID-19 deaths had been reported worldwide since January 22, 2020.
A total of 8,432 cases and 813 deaths were reported for SARS from March 17 to July 11, 2003.
SARS-CoV-1 was officially contained on July 5, 2003, within 9 months of its appearance (730).
In contrast, SARS-CoV-2 remains a significant global threat nearly two years after its emergence.
COVID-19 data are from the COVID-19 Data Repository by the Center for Systems Science and Engineering at Johns Hopkins University (731, 732).
SARS data are from the WHO (733) and were obtained from a dataset on GitHub (734).
See https://greenelab.github.io/covid19-review/ for the most recent version of this figure, which is updated daily.
At first, SARS-CoV-2’s rapid shift from an unknown virus to a significant worldwide threat closely paralleled the emergence of Severe acute respiratory syndrome-related coronavirus (SARS-CoV-1), which was responsible for the 2002-03 SARS epidemic.
The first documented case of COVID-19 was reported in Wuhan, China in November 2019, and the disease quickly spread worldwide in the early months of 2020.
In comparison, the first case of SARS was reported in November 2002 in the Guangdong Province of China, and it spread within China and then into several countries across continents during the first half of 2003 (250, 340, 730).
In fact, genome sequencing quickly revealed the virus causing COVID-19 to be a novel betacoronavirus closely related to SARS-CoV-1 (11).
While similarities between these two viruses are unsurprising given their close phylogenetic relationship, there are also some differences in how the viruses affect humans.
SARS-CoV-1 infection is severe, with an estimated case fatality rate (CFR) for SARS of 9.5% (340), while estimates of the CFR associated with COVID-19 are much lower, at up to 2% (1).
SARS-CoV-1 is highly contagious and spread primarily by droplet transmission, with a basic reproduction number (R0) of 4 (i.e., each person infected was estimated to infect four other people) (340).
There is still some controversy whether SARS-CoV-2 is primarily spread by droplets or is primarily airborne (258, 259, 262, 735).
Most estimates of its R0 fall between 2.5 and 3 (1).
Therefore, SARS is thought to be a deadlier and more transmissible disease than COVID-19.
With the 17-year difference between these two outbreaks, there were major differences in the tools available to efforts to organize international responses.
At the time that SARS-CoV-1 emerged, no new HCoV had been identified in almost 40 years (250).
The identity of the virus underlying the SARS disease remained unknown until April of 2003, when the SARS-CoV-1 virus was characterized through a worldwide scientific effort spearheaded by the World Health Organization (WHO) (250).
In contrast, the SARS-CoV-2 genomic sequence was released on January 3, 2020 (11), only days after the international community became aware of the novel pneumonia-like illness now known as COVID-19.
While SARS-CoV-1 belonged to a distinct lineage from the two other HCoVs known at the time of its discovery (340), SARS-CoV-2 is closely related to SARS-CoV-1 and is a more distant relative of another HCoV characterized in 2012, Middle East respiratory syndrome-related coronavirus(19, 736).
Significant efforts had been dedicated towards understanding SARS-CoV-1 and MERS-CoV and how they interact with human hosts.
Therefore, SARS-CoV-2 emerged under very different circumstances than SARS-CoV-1 in terms of scientific knowledge about HCoVs and the tools available to characterize them.
Despite the apparent advantages for responding to SARS-CoV-2 infections, COVID-19 has caused many orders of magnitude more deaths than SARS did (Figure 3).
The SARS outbreak was officially determined to be under control in July 2003, with the success credited to infection management practices such as mask wearing (250).
Middle East respiratory syndrome-related coronavirus (MERS-CoV) is still circulating and remains a concern; although the fatality rate is very high at almost 35%, the disease is much less easily transmitted, as its R0 has been estimated to be 1 (340).
The low R0 in combination with public health practices allowed for its spread to be contained (340).
Neither of these trajectories are comparable to that of SARS-CoV-2, which remains a serious threat worldwide over a year and a half after the first cases of COVID-19 emerged (Figure 3).
4.3.2 Potential Approaches to the Treatment of COVID-19
Therapeutic interventions can utilize two approaches: they can either mitigate the effects of an infection that harms an infected person, or they can hinder the spread of infection within a host by disrupting the viral life cycle.
The goal of the former strategy is to reduce the severity and risks of an active infection, while for the latter, it is to inhibit the replication of a virus once an individual is infected, potentially freezing disease progression.
Additionally, two major approaches can be used to identify interventions that might be relevant to managing an emerging disease or a novel virus: drug repurposing and drug development.
Drug repurposing involves identifying an existing compound that may provide benefits in the context of interest (737).
This strategy can focus on either approved or investigational drugs, for which there may be applicable preclinical or safety information (737).
Drug development, on the other hand, provides an opportunity to identify or develop a compound specifically relevant to a particular need, but it is often a lengthy and expensive process characterized by repeated failure (738).
Drug repurposing therefore tends to be emphasized in a situation like the COVID-19 pandemic due to the potential for a more rapid response.
Even from the early months of the pandemic, studies began releasing results from analyses of approved and investigational drugs in the context of COVID-19.
The rapid timescale of this response meant that, initially, most evidence came from observational studies, which compare groups of patients who did and did not receive a treatment to determine whether it may have had an effect.
This type of study can be conducted rapidly but is subject to confounding.
In contrast, randomized controlled trials (RCTs) are the gold-standard method for assessing the effects of an intervention.
Here, patients are prospectively and randomly assigned to treatment or control conditions, allowing for much stronger interpretations to be drawn; however, data from these trials take much longer to collect.
Both approaches have proven to be important sources of information in the development of a rapid response to the COVID-19 crisis, but as the pandemic draws on and more results become available from RCTs, more definitive answers are becoming available about proposed therapeutics.
Interventional clinical trials are currently investigating or have investigated a large number of possible therapeutics and combinations of therapeutics for the treatment of COVID-19 (Figure 4).
Figure 4:COVID-19 clinical trials.
Trials data are from the University of Oxford Evidence-Based Medicine Data Lab’s COVID-19 TrialsTracker (739).
As of December 31, 2020, there were 6,987 COVID-19 clinical trials of which 3,962 were interventional.
The study types include only types used in at least five trials.
Only interventional trials are analyzed in the figures depicting status, phase, and intervention.
Of the interventional trials, 98 trials had reported results as of December 31, 2020.
Recruitment status and trial phase are shown only for interventional trials in which the status or phase is recorded.
Common interventions refers to interventions used in at least ten trials.
Combinations of interventions, such as hydroxychloroquine with azithromycin, are tallied separately from the individual interventions.
See https://greenelab.github.io/covid19-review/ for the most recent version of this figure, which is updated daily.
The purpose of this review is to provide an evolving resource tracking the status of efforts to repurpose and develop drugs for the treatment of COVID-19.
We highlight four strategies that provide different paradigms for the identification of potential pharmaceutical treatments.
The WHO guidelines (81) and a systematic review (740) are complementary living documents that summarize COVID-19 therapeutics.
4.4 Repurposing Drugs for Symptom Management
A variety of symptom profiles with a range of severity are associated with COVID-19 (1).
In many cases, COVID-19 is not life threatening.
A study of COVID-19 patients in a hospital in Berlin, Germany reported that the highest risk of death was associated with infection-related symptoms, such as sepsis, respiratory symptoms such as ARDS, and cardiovascular failure or pulmonary embolism (741).
Similarly, an analysis in Wuhan, China reported that respiratory failure (associated with ARDS) and sepsis/multi-organ failure accounted for 69.5% and 28.0% of deaths, respectively, among 82 deceased patients (742).
COVID-19 is characterized by two phases.
The first is the acute response, where an adaptive immune response to the virus is established and in many cases can mitigate viral damage to organs (743).
The second phase characterizes more severe cases of COVID-19.
Here, patients experience a cytokine storm, whereby excessive production of cytokines floods into circulation, leading to systemic inflammation, immune dysregulation, and multiorgan dysfunction that can cause multiorgan failure and death if untreated (744).
ARDS-associated respiratory failure can occur during this phase.
Cytokine dysregulation was also identified in patients with SARS (745, 746).
In the early days of the COVID-19 pandemic, physicians sought to identify potential treatments that could benefit patients, and in some cases shared their experiences and advice with the medical community on social media sites such as Twitter (747).
These on-the-ground treatment strategies could later be analyzed retrospectively in observational studies or investigated in an interventional paradigm through RCTs.
Several notable cases involved the use of small-molecule drugs, which are synthesized compounds of low molecular weight, typically less than 1 kilodalton (kDa) (748).
Small-molecule pharmaceutical agents have been a backbone of drug development since the discovery of penicillin in the early twentieth century (749).
It and other antibiotics have long been among the best known applications of small molecules to therapeutics, but biotechnological developments such as the prediction of protein-protein interactions (PPIs) have facilitated advances in precise targeting of specific structures using small molecules (749).
Small molecule drugs today encompass a wide range of therapeutics beyond antibiotics, including antivirals, protein inhibitors, and many broad-spectrum pharmaceuticals.
Many treatments considered for COVID-19 have relied on a broad-spectrum approach.
These treatments do not specifically target a virus or particular host receptor, but rather induce broad shifts in host biology that are hypothesized to be potential inhibitors of the virus.
This approach relies on the fact that when a virus enters a host, the host becomes the virus’s environment.
Therefore, the state of the host can also influence the virus’s ability to replicate and spread.
The administration and assessment of broad-spectrum small-molecule drugs on a rapid time course was feasible because they are often either available in hospitals, or in some cases may also be prescribed to a large number of out-patients.
One of the other advantages is that these well-established compounds, if found to be beneficial, are often widely available, in contrast to boutique experimental drugs.
In some cases, prior data was available from experiments examining the response of other HCoVs or HCoV infections to a candidate drug.
In addition to non-pharmaceutical interventions such as encouraging non-intubated patients to adopt a prone position (750), knowledge about interactions between HCoVs and the human body, many of which emerged from SARS and MERS research over the past two decades, led to the suggestion that a number of common drugs might benefit COVID-19 patients.
However, the short duration and low case numbers of prior outbreaks were less well-suited to the large-scale study of clinical applications than the COVID-19 pandemic is.
As a result, COVID-19 has presented the first opportunity to robustly evaluate treatments that were common during prior HCoV outbreaks to determine their clinical efficacy.
The first year of the COVID-19 pandemic demonstrated that there are several different trajectories that these clinically suggested, widely available candidates can follow when assessed against a widespread, novel viral threat.
One approach to identifying candidate small molecule drugs was to look at the approaches used to treat SARS and MERS.
Treatment of SARS and MERS patients prioritized supportive care and symptom management (340).
Among the clinical treatments for SARS and MERS that were explored, there was generally a lack of evidence indicating whether they were effective.
Most of the supportive treatments for SARS were found inconclusive in meta-analysis (751), and a 2004 review reported that not enough evidence was available to make conclusions about most treatments (752).
However, one strategy adopted from prior HCoV outbreaks is currently the best-known treatment for severe cases of COVID-19.
Corticosteroids represent broad-spectrum treatments and are a well-known, widely available treatment for pneumonia (753–758) that have also been debated as a possible treatment for ARDS (759–764).
Corticosteroids were also used and subsequently evaluated as possible supportive care for SARS and MERS.
In general, studies and meta-analyses did not identify support for corticosteroids to prevent mortality in these HCoV infections (765–767); however, one found that the effects might be masked by variability in treatment protocols, such as dosage and timing (752).
While the corticosteroids most often used to treat SARS were methylprednisolone and hydrocortisone, availability issues for these drugs at the time led to dexamethasone also being used in North America (768).
Dexamethasone (9α-fluoro-16α-methylprednisolone) is a synthetic corticosteroid that binds to glucocorticoid receptors (769, 770).
It functions as an anti-inflammatory agent by binding to glucocorticoid receptors with higher affinity than endogenous cortisol (771).
Dexamethasone and other steroids are widely available and affordable, and they are often used to treat community-acquired pneumonia (772) as well as chronic inflammatory conditions such as asthma, allergies, and rheumatoid arthritis (773–775).
Immunosuppressive drugs such as steroids are typically contraindicated in the setting of infection (776), but because COVID-19 results in hyperinflammation that appears to contribute to mortality via lung damage, immunosuppression may be a helpful approach to treatment (158).
A clinical trial that began in 2012 recently reported that dexamethasone may improve outcomes for patients with ARDS (759), but a meta-analysis of a small amount of available data about dexamethasone as a treatment for SARS suggested that it may, in fact, be associated with patient harm (777).
However, the findings in SARS may have been biased by the fact that all of the studies examined were observational and a large number of inconclusive studies were not included (778).
The questions of whether and when to counter hyperinflammation with immunosuppression in the setting of COVID-19 (as in SARS (746)) was an area of intense debate, as the risks of inhibiting antiviral immunity needed to be weighed against the beneficial anti-inflammatory effects (779).
As a result, guidelines early in the pandemic typically recommended avoiding treating COVID-19 patients with corticosteroids such as dexamethasone (777).
Despite this initial concern, dexamethasone was evaluated as a potential treatment for COVID-19 (Appendix 1).
Dexamethasone treatment comprised one arm of the multi-site Randomized Evaluation of COVID-19 Therapy (RECOVERY) trial in the United Kingdom (780).
This study found that the 28-day mortality rate was lower in patients receiving dexamethasone than in those receiving standard of care (SOC).
However, this finding was driven by differences in mortality among patients who were receiving mechanical ventilation or supplementary oxygen at the start of the study.
The report indicated that dexamethasone reduced 28-day mortality relative to SOC in patients who were ventilated (29.3% versus 41.4%) and among those who were receiving oxygen supplementation (23.3% versus 26.2%) at randomization, but not in patients who were breathing independently (17.8% versus 14.0%).
These findings also suggested that dexamethasone may have reduced progression to mechanical ventilation, especially among patients who were receiving oxygen support at randomization.
Other analyses have supported the importance of disease course in determining the efficacy of dexamethasone: additional results suggest greater potential for patients who have experienced symptoms for at least seven days and patients who were not breathing independently (781).
A meta-analysis that evaluated the results of the RECOVERY trial alongside trials of other corticosteroids, such as hydrocortisone, similarly concluded that corticosteroids may be beneficial to patients with severe COVID-19 who are receiving oxygen supplementation (782).
Thus, it seems likely that dexamethasone is useful for treating inflammation associated with immunopathy or cytokine release syndrome (CRS), which is a condition caused by detrimental overactivation of the immune system (1).
In fact, corticosteroids such as dexamethasone are sometimes used to treat CRS (783).
Guidelines were quickly updated to encourage the use of dexamethasone in severe cases (784), and this affordable and widely available treatment rapidly became a valuable tool against COVID-19 (785), with demand surging within days of the preprint’s release (786).
4.5 Approaches Targeting the Virus
Therapeutics that directly target the virus itself hold the potential to prevent people infected with SARS-CoV-2 from developing potentially damaging symptoms (Figure 5).
Such drugs typically fall into the broad category of antivirals.
Antiviral therapies hinder the spread of a virus within the host, rather than destroying existing copies of the virus, and these drugs can vary in their specificity to a narrow or broad range of viral targets.
This process requires inhibiting the replication cycle of a virus by disrupting one of six fundamental steps (787).
In the first of these steps, the virus attaches to and enters the host cell through endocytosis.
Then the virus undergoes uncoating, which is classically defined as the release of viral contents into the host cell.
Next, the viral genetic material enters the nucleus where it gets replicated during the biosynthesis stage.
During the assembly stage, viral proteins are translated, allowing new viral particles to be assembled.
In the final step new viruses are released into the extracellular environment.
Although antivirals are designed to target a virus, they can also impact other processes in the host and may have unintended effects.
Therefore, these therapeutics must be evaluated for both efficacy and safety.
As the technology to respond to emerging viral threats has also evolved over the past two decades, a number of candidate treatments have been identified for prior viruses that may be relevant to the treatment of COVID-19.
Figure 5:Mechanisms of Action for Potential Therapeutics
Potential therapeutics currently being studied can target the SARS-CoV-2 virus or modify the host environment through many different mechanisms.
Here, the relationships between the virus, host cells, and several therapeutics are visualized.
Drug names are color-coded according to the grade assigned to them by the Center for Cytokine Storm Treatment & Laboratory’s CORONA Project (788) (Green = A, Lime = B, Orange = C, and Red = D).
Many antiviral drugs are designed to inhibit the replication of viral genetic material during the biosynthesis step.
Unlike DNA viruses, which can use the host enzymes to propagate themselves, RNA viruses like SARS-CoV-2 depend on their own polymerase, the RNA-dependent RNA polymerase (RdRP), for replication (789, 790).
RdRP is therefore a potential target for antivirals against RNA viruses.
Disruption of RdRP is the proposed mechanism underlying the treatment of SARS and MERS with ribavirin (791).
Ribavirin is an antiviral drug effective against other viral infections that was often used in combination with corticosteroids and sometimes interferon (IFN) medications to treat SARS and MERS (250).
However, analyses of its effects in retrospective and in vitro analyses of SARS and the SARS-CoV-1 virus, respectively, have been inconclusive (250).
While IFNs and ribavirin have shown promise in in vitro analyses of MERS, their clinical effectiveness remains unknown (250).
The current COVID-19 pandemic has provided an opportunity to assess the clinical effects of these treatments.
As one example, ribivarin was also used in the early days of COVID-19, but a retrospective cohort study comparing patients who did and did not receive ribivarin revealed no effect on the mortality rate (792).
Since nucleotides and nucleosides are the natural building blocks for RNA synthesis, an alternative approach has been to explore nucleoside and nucleotide analogs for their potential to inhibit viral replication.
Analogs containing modifications to nucleotides or nucleosides can disrupt key processes including replication (793).
A single incorporation does not influence RNA transcription; however, multiple events of incorporation lead to the arrest of RNA synthesis (794).
One candidate antiviral considered for the treatment of COVID-19 is favipiravir (Avigan), also known as T-705, which was discovered by Toyama Chemical Co., Ltd. (795).
It was previously found to be effective at blocking viral amplification in several influenza subtypes as well as other RNA viruses, such as Flaviviridae and Picornaviridae, through a reduction in plaque formation (796) and viral replication in Madin-Darby canine kidney cells (797).
Favipiravir (6-fluoro-3-hydroxy-2-pyrazinecarboxamide) acts as a purine and purine nucleoside analogue that inhibits viral RNA polymerase in a dose-dependent manner across a range of RNA viruses, including influenza viruses (798–802).
Biochemical experiments showed that favipiravir was recognized as a purine nucleoside analogue and incorporated into the viral RNA template.
In 2014, the drug was approved in Japan for the treatment of influenza that was resistant to conventional treatments like neuraminidase inhibitors (803).
Though initial analyses of favipiravir in observational studies of its effects on COVID-19 patients were promising, recent results of two small RCTs suggest that it is unlikely to affect COVID-19 outcomes (Appendix 1).
In contrast, another nucleoside analog, remdesivir, is one of the few treatments against COVID-19 that has received FDA approval.
Remdesivir (GS-5734) is an intravenous antiviral that was proposed by Gilead Sciences as a possible treatment for Ebola virus disease.
It is metabolized to GS-441524, an adenosine analog that inhibits a broad range of polymerases and then evades exonuclease repair, causing chain termination (804–806).
Gilead received an emergency use authorization (EUA) for remdesivir from the FDA early in the pandemic (May 2020) and was later found to reduce mortality and recovery time in a double-blind, placebo-controlled, phase III clinical trial performed at 60 trial sites, 45 of which were in the United States (807–810).
Subsequently, the WHO Solidarity trial, a large-scale, open-label trial enrolling 11,330 adult in-patients at 405 hospitals in 30 countries around the world, reported no effect of remdesivir on in-hospital mortality, duration of hospitalization, or progression to mechanical ventilation (811).
Therefore, additional clinical trials of remdesivir in different patient pools and in combination with other therapies may be needed to refine its use in the clinic and determine the forces driving these differing results.
Remdesivir offers proof of principle that SARS-CoV-2 can be targeted at the level of viral replication, since remdesivir targets the viral RNA polymerase at high potency.
Identification of such candidates depends on knowledge about the virological properties of a novel threat.
However, the success and relative lack of success, respectively, of remdesivir and favipiravir underscore the fact that drugs with similar mechanisms will not always produce similar results in clinical trials.
4.6 Disrupting Host-Virus Interactions
4.6.1 Interrupting Viral Colonization of Cells
Some of the most widely publicized examples of efforts to repurpose drugs for COVID-19 are broad-spectrum, small-molecule drugs where the mechanism of action made it seem that the drug might disrupt interactions between SARS-CoV-2 and human host cells (Figure 5).
However, the exact outcomes of such treatments are difficult to predict a priori, and there are several examples where early enthusiasm was not borne out in subsequent trials.
One of the most famous examples of an analysis of whether a well-known medication could provide benefits to COVID-19 patients came from the assessment of chloroquine (CQ) and hydroxychloroquine (HCQ), which are used for the treatment and prophylaxis of malaria as well as the treatment of lupus erythematosus and rheumatoid arthritis in adults (812).
These drugs are lysosomotropic agents, meaning they are weak bases that can pass through the plasma membrane.
It was thought that they might provide benefits against SARS-CoV-2 by interfering with the digestion of antigens within the lysosome and inhibiting CD4 T-cell stimulation while promoting the stimulation of CD8 T-cells (813).
These compounds also have anti-inflammatory properties (813) and can decrease the production of certain key cytokines involved in the immune response, including interleukin-6 (IL-6) and inhibit the stimulation of Toll-like receptors (TLR) and TLR signaling (813).
In vitro analyses reported that CQ inhibited cell entry of SARS-CoV-1 (814) and that both CQ and HCQ inhibited viral replication within cultured cells (815), leading to early hope that it might provide similar therapeutic or protective effects in patients.
However, while the first publication on the clinical application of these compounds to the inpatient treatment of COVID-19 was very positive (816), it was quickly discredited (817).
Over the following months, extensive evidence emerged demonstrating that CQ and HCQ offered no benefits for COVID-19 patients and, in fact, carried the risk of dangerous side effects (Appendix 1).
The nail in the coffin came when findings from the large-scale RECOVERY trial were released on October 8, 2020.
This study enrolled 11,197 hospitalized patients whose physicians believed it would not harm them to participate and used a randomized, open-label design to study the effects of HCQ compared to standard of care (SOC) at 176 hospitals in the United Kingdom (818).
Rates of COVID-19-related mortality did not differ between the control and HCQ arms, but patients receiving HCQ were slightly more likely to die due to cardiac events.
Patients who received HCQ also had a longer duration of hospitalization than patients receiving usual care and were more likely to progress to mechanical ventilation or death (as a combined outcome).
As a result, enrollment in the HCQ arm of the RECOVERY trial was terminated early (819).
The story of CQ/HCQ therefore illustrates how initial promising in vitro analyses can fail to translate to clinical usefulness.
A similar story has arisen with the broad-spectrum, small-molecule anthelmintic ivermectin, which is a synthetic analog of avermectin, a bioactive compound produced by a microorganism known as Streptomyces avermectinius and Streptomyces avermitilis(820, 821).
Avermectin disrupts the ability of parasites to avoid the host immune response by blocking glutamate-gated chloride ion channels in the peripheral nervous system from closing, leading to hyperpolarization of neuronal membranes, disruption of neural transmission, and paralysis (820, 822, 823).
Ivermectin has been used since the early 1980s to treat endo- and ecto-parasitic infections by helminths, insects, and arachnids in veterinary contexts (820, 824) and since the late 1980s to treat human parasitic infections as well (820, 822).
More recent research has indicated that ivermectin might function as a broad-spectrum antiviral by disrupting the trafficking of viral proteins by both RNA and DNA viruses (823, 825, 826), although most of these studies have demonstrated this effect in vitro(826).
The potential for antiviral effects on SARS-CoV-2 were investigated in vitro, and ivermectin was found to inhibit viral replication in a cell line derived from Vero cells (Vero-hSLAM) (827).
However, inhibition of viral replication was achieved at concentrations that were much higher than that explored by existing dosage guidelines (828, 829), which are likely to be associated with significant side effects due to the increased potential that the compound could cross the mammalian blood-brain barrier (830, 831).
Retrospective studies and small RCTs began investigating the effects of standard doses of this low-cost, widely available drug.
One retrospective study reported that ivermectin reduced all-cause mortality (832) while another reported no difference in clinical outcomes or viral clearance (833).
Small RCTs enrolling less than 50 patients per arm have also reported a wide array of positive (834–838) and negative results (839, 840).
A slightly larger RCT enrolling 115 patients in two arms reported inconclusive results (841).
Hope for the potential of ivermectin peaked with the release of a preprint reporting results of a multicenter, double-blind RCT where a four-day course of ivermectin was associated with clinical improvement and earlier viral clearance in 400 symptomatic patients and 200 close contacts (842); however, concerns were raised about both the integrity of the data and the paper itself (843, 844), and this study was removed by the preprint server Research Square (845).
A similarly sized RCT suggested no effect on the duration of symptoms among 400 patients split evenly across the intervention and control arms (846), and although meta-analyses have reported both null (847, 848) and beneficial (849–856) effects of ivermectin on COVID-19 outcomes, the certainty is likely to be low (850).
These findings are potentially biased by a small number of low-quality studies, including the preprint that has been taken down (857), and the authors of one (858) have issued a notice (849) that they will revise their study with the withdrawn study removed.
Thus, much like HCQ/CQ, enthusiasm for research that either has not or should not have passed peer review has led to large numbers of patients worldwide receiving treatments that might not have any effect or could even be harmful.
Additionally, comments on the now-removed preprint include inquiries into how best to self-administer veterinary ivermectin as a prophylactic (845), and the FDA has posted information explaining why veterinary ivermectin should not be taken by humans concerned about COVID-19 (859).
Ivermectin is now one of several candidate therapeutics being investigated in the large-scale TOGETHER (860) and PRINCIPLE (861) clinical trials.
The TOGETHER trial, which previously demonstrated no effect of HCQ and lopinavir-ritonavir (862), released preliminary results in early August 2021 suggesting that ivermectin also has no effect on COVID-19 outcomes (863).
While CQ/HCQ and ivermectin are well-known medications that have long been prescribed in certain contexts, investigation of another well-established type of pharmaceutical was facilitated by the fact that it was already being taken by a large number of COVID-19 patients.
Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin II receptor blockers (ARBs) are among today’s most commonly prescribed medications, often being used to control blood pressure (864, 865).
In the United States, for example, they are prescribed well over 100,000,000 times annually (866).
Prior to the COVID-19 pandemic, the relationship between ACE2, ACEIs, and SARS had been considered as possible evidence that ACE2 could serve as a therapeutic target (867), and the connection had been explored through in vitro and molecular docking analysis (868) but ultimately was not pursued clinically (869).
Data from some animal models suggest that several, but not all, ACEIs and several ARBs increase ACE2 expression in the cells of some organs (870), but clinical studies have not established whether plasma ACE2 expression is increased in humans treated with these medications (871).
In this case, rather than introducing ARBs/ACEIs, a number of analyses have investigated whether discontinuing use affects COVID-19 outcomes.
An initial observational study of the association of exposure to ACEIs or ARBs with outcomes in COVID-19 was retracted from the New England Journal of Medicine(872) due to concerns related to data availability (873).
As RCTs have become available, they have demonstrated no effect of continuing versus discontinuing ARBs/ACEIs on patient outcomes (874, 875) (Appendix 1).
Thus, once again, despite a potential mechanistic association with the pathology of SARS-CoV-2 infection, these medications were not found to influence the trajectory of COVID-19 illness.
For medications that are widely known and common, clinical research into their efficacy against a novel threat can be developed very quickly.
This feasibility can present a double-edged sword.
For example, HCQ and CQ were incorporated into SOC in many countries early in the pandemic and had to be discontinued once their potential to harm COVID-19 patients became apparent (876, 877).
Dexamethasone remains the major success story from this category of repurposed drugs and is likely to have saved a large number of lives since summer 2020 (785).
4.6.2 Manipulating the Host Immune Response
Treatments based on understanding a virus and/or how a virus interacts with the human immune system can fall into two categories: they can interact with the innate immune response, which is likely to be a similar response across viruses, or they can be specifically designed to imitate the adaptive immune response to a particular virus.
In the latter case, conservation of structure or behavior across viruses enables exploring whether drugs developed for one virus can treat another.
During the COVID-19 pandemic, a number of candidate therapeutics have been explored in these categories, with varied success.
Knowledge gained from trying to understand SARS-CoV-1 and MERS-CoV from a fundamental biological perspective and characterize how they interact with the human immune system provides a theoretical basis for identifying candidate therapies.
Biologics are a particularly important class of drugs for efforts to address HCoV through this paradigm.
They are produced from components of living organisms or viruses, historically primarily from animal tissues, but have become increasingly feasible to produce as recombinant technologies have advanced (878).
There are many differences on the development side between biologics and synthesized pharmaceuticals, such as small molecule drugs.
Typically, biologics are orders of magnitude larger than small molecule drugs and are catabolized by the body to their amino acid components (879).
They are often heat sensitive, and their toxicity can vary, as it is not directly associated with the primary effects of the drug; in general, their physiochemical properties are much less understood compared to small molecules (879).
Biologics include significant medical breakthroughs such as insulin for the management of diabetes and vaccines, as well monoclonal antibodies (mAbs) and interferons (IFNs), which can be used to target the host immune response after infection.
mAbs have revolutionized the way we treat human diseases and have become some of the best-selling drugs in the pharmaceutical market in recent years (880).
There are currently 79 FDA approved mAbs on the market, including antibodies for viral infections (e.g. Ibalizumab for Human immunodeficiency virus and Palivizumab for Respiratory syncytial virus) (880, 881).
Virus-specific neutralizing antibodies commonly target viral surface glycoproteins or host structures, thereby inhibiting viral entry through receptor binding interference (882, 883).
This interference is predicted to reduce the viral load, mitigate disease, and reduce overall hospitalization.
mAbs can be designed for a particular virus, and significant advances have been made in the speed at which new mAbs can be identified and produced.
At the time of the SARS and MERS epidemics, interest in mAbs to reduce infection was never realized (884, 885), but this allowed for mAbs to quickly be considered among the top candidates against COVID-19.
4.6.2.1 Biologics and the Innate Immune Response
Deaths from COVID-19 often occur when inflammation becomes dysregulated following an immune response to the SARS-CoV-2 virus.
Therefore, one potential approach to reducing COVID-19 mortality rates is to manage the inflammatory response in severely ill patients.
One candidate therapeutic identified that uses this mechanism is tocilizumab (TCZ).
TCZ is a mAb that was developed to manage chronic inflammation caused by the continuous synthesis of the cytokine IL-6 (886).
IL-6 is a pro-inflammatory cytokine belonging to the interleukin family, which is comprised by immune system regulators that are primarily responsible for immune cell differentiation.
Often used to treat chronic inflammatory conditions such as rheumatoid arthritis (886), TCZ has become a pharmaceutical of interest for the treatment of COVID-19 because of the role IL-6 plays in this disease.
It has also been approved to treat CRS caused by CAR-T treatments (887).
While the secretion of IL-6 can be associated with chronic conditions, IL-6 is a key player in the innate immune response and is secreted by macrophages in response to the detection of pathogen-associated molecular patterns and damage-associated molecular patterns (886).
An analysis of 191 in-patients at two Wuhan hospitals revealed that blood concentrations of IL-6 differed between patients who did and did not recover from COVID-19.
Patients who ultimately died had higher IL-6 levels at admission than those who recovered (83).
Additionally, IL-6 levels remained higher throughout the course of hospitalization in the patients who ultimately died (83).
Currently, TCZ is being administered either as a monotherapy or in combination with other treatments in 73 interventional COVID-19 clinical trials (Figure 4).
A number of retrospective studies have been conducted in several countries (888–893).
In general, these studies have reported a positive effect of TCZ on reducing mortality in COVID-19 patients, although due to their retrospective designs, significant limitations are present in all of them (Appendix 1).
It was not until February 11, 2021 that a preprint describing preliminary results of the first RCT of TCZ was released as part of the RECOVERY trial (894).
TCZ was found to reduce 28-day mortality from 33% in patients receiving SOC alone to 29% in those receiving TCZ.
Combined analysis of the RECOVERY trial data with data from smaller RCTs suggested a 13% reduction in 28-day mortality (894).
While this initial report did not include the full results expected from the RECOVERY trial, this large-scale, RCT provides strong evidence that TCZ may offer benefits for COVID-19 patients.
The RECOVERY trial along with results from several other RCTs (895–899) were cited as support for the EUA issued for TCZ in June 2021 (900).
However, the fact that TCZ suppresses the immune response means that it does carry risks for patients, especially a potential risk of secondary infection (Appendix 1).
TCZ is just one example of a candidate drug targeting the host immune response and specifically excessive inflammation.
For example, interferons (IFNs) have also been investigated; these are a family of cytokines critical to activating the innate immune response against viral infections.
Synairgen has been investigating a candidate drug, SNG001, which is an IFN-𝛽-1a formulation to be delivered to the lungs via inhalation (901) that they reported reduced progression to ventilation in a double-blind, placebo-controlled, multi-center study of 101 patients with an average age in the late 50s (902, 903).
However, these findings were not supported by the large-scale WHO Solidarity trial, which reported no significant effect of IFN-β-1a on patient survival during hospitalization (811), although differences in the designs of the two studies, and specifically the severity of illness among enrolled patients, may have influenced their divergent outcomes (Appendix 1).
Other biologics influencing inflammation are also being explored (Appendix 1).
It is also important that studies focused on inflammation as a possible therapeutic target consider the potential differences in baseline inflammation among patients from different backgrounds, which may be caused by differing life experiences (see (339)).
4.6.2.2 Biologics and the Adaptive Immune Response
While TCZ is an example of an mAb focused on managing the innate immune response, other treatments are more specific, targeting the adaptive immune response after an infection.
In some cases, treatments can utilize biologics obtained directly from recovered individuals.
From the very early days of the COVID-19 pandemic, polyclonal antibodies from convalescent plasma were investigated as a potential treatment for COVID-19 (904, 905).
Convalescent plasma was used in prior epidemics including SARS, Ebola Virus Disease, and even the 1918 Spanish Influenza (904, 906).
Use of convalescent plasma transfusion (CPT) over more than a century has aimed to reduce symptoms and improve mortality in infected people (906), possibly by accelerating viral clearance (904).
However, it seems unlikely that this classic treatment confers any benefit for COVID-19 patients.
Several systematic reviews have investigated whether CPT reduced mortality in COVID-19 patients, and although findings from early in the pandemic (up to April 19, 2020) did support use of CPT (906), the tide has shifted as the body of available literature has grown (907).
While titer levels were suggested as a possible determining factor in the success of CPT against COVID-19 (908), the large-scale RECOVERY trial evaluated the effect of administering high-titer plasma specifically and found no effect on mortality or hospital discharge over a 28-day period (909).
These results thus suggest that, despite initial optimism and an EUA from the FDA, CPT is unlikely to be an effective therapeutic for COVID-19.
A different narrative is shaping up around the use of mAbs specifically targeting SARS-CoV-2.
During the first SARS epidemic in 2002, neutralizing antibodies (nAbs) were found in SARS-CoV-1-infected patients (910, 911).
Several studies following up on these findings identified various S-glycoprotein epitopes as the major targets of nAbs against SARS-CoV-1 (912).
Coronaviruses use trimeric spike (S) glycoproteins on their surface to bind to the host cell, allowing for cell entry (25, 33).
Each S glycoprotein protomer is comprised of an S1 domain, also called the receptor binding domain (RBD), and an S2 domain.
The S1 domain binds to the host cell while the S2 domain facilitates the fusion between the viral envelope and host cell membranes (912).
The genomic identity between the RBD of SARS-CoV-1 and SARS-CoV-2 is around 74% (913).
Due to this high degree of similarity, preexisting antibodies against SARS-CoV-1 were initially considered candidates for neutralizing activity against SARS-CoV-2.
While some antibodies developed against the SARS-CoV-1 spike protein showed cross-neutralization activity with SARS-CoV-2 (914, 915), others failed to bind to SARS-CoV-2 spike protein at relevant concentrations (16).
Cross-neutralizing activities were dependent on whether the epitope recognized by the antibodies were conserved between SARS-CoV-1 and SARS-CoV-2 (914).
Technological advances in antibody drug design as well as in structural biology massively accelerated the discovery of novel antibody candidates and the mechanisms by which they interact with the target structure.
Within just a year of the structure of the SARS-CoV-2 spike protein being published, an impressive pipeline of monoclonal antibodies targeting SARS-CoV-2 entered clinical trials, with hundreds more candidates in preclinical stages.
The first human monoclonal neutralizing antibody specifically against the SARS-CoV-2 S glycoprotein was developed using hybridoma technology (916), where antibody-producing B-cells developed by mice are inserted into myeloma cells to produce a hybrid cell line (the hybridoma) that is grown in culture.
The 47D11 antibody clone was able to cross-neutralize SARS-CoV-1 and SARS-CoV-2.
This antibody (now ABVV-47D11) has recently entered clinical trials in collaboration with AbbVie.
Additionally, an extensive monoclonal neutralizing antibody pipeline has been developed to combat the ongoing pandemic, with over 50 different antibodies in clinical trials (917).
Thus far, the monotherapy sotrovimab and two antibody cocktails (bamlanivimab/estesevimab and casirivimab/imdevimab) have been granted EUAs by the FDA.
One of the studied antibody cocktails consists of bamlanivimab and estesevimab.
Bamlanivimab (Ly-CoV555) is a human mAb that was derived from convalescent plasma donated by a recovered COVID-19 patient, evaluated in research by the National Institute of Allergy and Infectious Diseases (NIAID), and subsequently developed by AbCellera and Eli Lilly.
The neutralizing activity of bamlanivimab was initially demonstrated in vivo using a nonhuman primate model (918).
Based on these positive preclinical data, Eli Lilly initiated the first human clinical trial for a monoclonal antibody against SARS-CoV-2.
The phase 1 trial, which was conducted in hospitalized COVID-19 patients, was completed in August 2020 (919).
Estesevimab (LY-CoV016 or JS-016) is also a monoclonal neutralizing antibody against the spike protein of SARS-CoV-2.
It was initially developed by Junshi Biosciences and later licensed and developed through Eli Lilly.
A phase 1 clinical trial to assess the safety of etesevimab was completed in October 2020 (920).
Etesevimab was shown to bind a different epitope on the spike protein than bamlanivimab, suggesting that the two antibodies used as a combination therapy would further enhance their clinical use compared to a monotherapy (921).
To assess the efficacy and safety of bamlanivimab alone or in combination with etesevimab for the treatment of COVID-19, a phase 2/3 trial (BLAZE-1) (922) was initiated.
The interim analysis of the phase 2 portion suggested that bamlanivimab alone was able to accelerate the reduction in viral load (923).
However, more recent data suggests that only the bamlanivimab/etesevimab combination therapy is able to reduce viral load in COVID-19 patients (921).
Based on this data, the combination therapy received an EUA for COVID-19 from the FDA in February 2021 (924).
A second therapy is comprised of casirivimab and imdevimab (REGN-COV2).
Casirivimab (REGN10933) and imdevimab (REGN10987) are two monoclonal antibodies against the SARS-CoV-2 spike protein.
They were both developed by Regeneron in a parallel high-throughput screening (HTS) to identify neutralizing antibodies from either humanized mice or patient-derived convalescent plasma (925).
In these efforts, multiple antibodies were characterized for their ability to bind and neutralize the SARS-CoV-2 spike protein.
The investigators hypothesized that an antibody cocktail, rather than each individual antibody, could increase the therapeutic efficacy while minimizing the risk for virus escape.
Therefore, the authors tested pairs of individual antibodies for their ability to simultaneously bind the RBD of the spike protein.
Based on this data, casirivimab and imdevimab were identified as the lead antibody pair, resulting in the initiation of two clinical trials (926, 927).
Data from this phase 1-3 trial published in the New England Journal of Medicine shows that the REGN-COV2 antibody cocktail reduced viral load, particularly in patients with high viral load or whose endogenous immune response had not yet been initiated (928).
However, in patients who already initiated an immune response, exogenous addition of REGN-COV2 did not improve the endogenous immune response.
Both doses were well tolerated with no serious events related to the antibody cocktail.
Based on this data, the FDA granted an EUA for REGN-COV2 in patients with mild to moderate COVID-19 who are at risk of developing severe disease (929).
Ongoing efforts are trying to evaluate the efficacy of REGN-COV2 to improve clinical outcomes in hospitalized patients (926).
Sotrovimab is the most recent mAb to receive an EUA.
It was identified in the memory B cells of a 2003 survivor of SARS (930) and was found to be cross-reactive with SARS-CoV-2 (915).
This cross-reactivity is likely attributable to conservation within the epitope, with 17 out of 22 residues conserved between the two viruses, four conservatively substituted, and one semi-conservatively substituted (915).
In fact, these residues are highly conserved among sarbecoviruses, a clade that includes SARS-CoV-1 and SARS-CoV-2 (915).
This versatility has led to it being characterized as a “super-antibody” (931), a potent, broadly neutralizing antibody (932).
Interim analysis of data from a clinical trial (933) reported high safety and efficacy of this mAb in 583 COVID-19 patients (934).
Compared to placebo, sotrovimab was found to be 85% more effective in reducing progression to the primary endpoint, which was the proportion of patients who, within 29 days, were either hospitalized for more than 24 hours or died.
Additionally, rates of adverse events were comparable, and in some cases lower, among patients receiving sotrovimab compared to patients receiving a placebo.
Sotrovimab therefore represents a mAb therapeutic that is effective against SARS-CoV-2 and may also be effective against other sarbecoviruses.
Several potential limitations remain in the application of mAbs to the treatment of COVID-19.
One of the biggest challenges is identifying antibodies that not only bind to their target, but also prove to be beneficial for disease management.
Currently, use of mAbs is limited to people with mild to moderate disease that are not hospitalized, and it has yet to be determined whether they can be used as a successful treatment option for severe COVID-19 patients.
While preventing people from developing severe illness provides significant benefits, patients with severe illness are at the greatest risk of death, and therefore therapeutics that provide benefits against severe illness are particularly desirable.
It remains to be seen whether mAbs confer any benefits for patients in this category.
Another concern about therapeutics designed to amplify the response to a specific viral target is that they may need to be modified as the virus evolves.
With the ongoing global spread of new SARS-CoV-2 variants, there is a growing concern that mutations in SARS-CoV-2 spike protein could escape antibody neutralization, thereby reducing the efficacy of monoclonal antibody therapeutics and vaccines.
A comprehensive mutagenesis screen recently identified several amino acid substitutions in the SARS-CoV-2 spike protein that can prevent antibody neutralization (935).
While some mutations result in resistance to only one antibody, others confer broad resistance to multiple mAbs as well as polyclonal human sera, suggesting that some amino acids are “hotspots” for antibody resistance.
However, it was not investigated whether the resistance mutations identified result in a fitness advantage.
Accordingly, an impact on neutralizing efficiency has been reported for the B.1.1.7 (Alpha) variant first identified in the UK and the B.1.351 (Beta) variant first identified in in South Africa (936–938).
As of June 25, 2021, the CDC recommended a pause in the use of bamlanivimab and etesevimab due to decreased efficacy against the P.1 (Gamma) and B.1.351 (Beta) variants of SARS-CoV-2 (939).
While the reported impact on antibody neutralization needs to be confirmed in vivo, it suggests that some adjustments to therapeutic antibody treatments may be necessary to maintain the efficacy that was reported in previous clinical trials.
Several strategies have been employed to try to mitigate the risk of diminished antibody neutralization.
Antibody cocktails such as those already holding an EUA may help overcome the risk for attenuation of the neutralizing activity of a single monoclonal antibody.
These cocktails consist of antibodies that recognize different epitopes on the spike protein, decreasing the likelihood that a single amino acid change can cause resistance to all antibodies in the cocktail.
However, neutralizing resistance can emerge even against an antibody cocktail if the individual antibodies target subdominant epitopes (937).
Another strategy is to develop broadly neutralizing antibodies that target structures that are highly conserved, as these are less likely to mutate (940, 941) or to target epitopes that are insensitive to mutations (942).
Sotrovimab, one such “super-antibody”, is thought to be somewhat robust to neutralization escape (943) and has been found to be effective against all variants assessed as of August 12, 2021 (944).
Another antibody (ADG-2) targets a highly conserved epitope that overlaps the hACE2 binding site of all clade 1 sarbecoviruses (945).
Prophylactic administration of ADG-2 in an immunocompetent mouse model of COVID-19 resulted in protection against viral replication in the lungs and respiratory burden.
Since the epitope targeted by ADG-2 represents an Achilles’ heel for clade 1 sarbecoviruses, this antibody, like sotrovimab, might be a promising candidate against all circulating variants as well as emerging SARS-related coronaviruses.
To date, it has fared well against the Alpha, Beta, Gamma, and Delta variants (944).
The development of mAbs against SARS-CoV-2 has made it clear that this technology is rapidly adaptable and offers great potential for the response to emerging viral threats.
However, additional investigation may be needed to adapt mAb treatments to SARS-CoV-2 as it evolves and potentially to pursue designs that confer benefits for patients at the greatest risk of death.
While polyclonal antibodies from convalescent plasma have been evaluated as a treatment for COVID-19, these studies have suggested fewer potential benefits against SARS-CoV-2 than mAbs; convalescent plasma therapy has been thoroughly reviewed elsewhere (904, 905).
Thus, advances in biologics for COVID-19 illustrate that an understanding of how the host and virus interact can guide therapeutic approaches.
The FDA authorization of two combination mAb therapies, in particular, underscores the potential for this strategy to allow for a rapid response to a novel pathogen.
Additionally, while TCZ is not yet as established, this therapy suggests that the strategy of using biologics to counteract the cytokine storm response may provide therapies for the highest-risk patients.
4.7 High-Throughput Screening for Drug Repurposing
The drug development process is slow and costly, and developing compounds specifically targeted to an emerging viral threat is not a practical short-term solution.
Screening existing drug compounds for alternative indications is a popular alternative (946–949).
HTS has been a goal of pharmaceutical development since at least the mid-1980s (950).
Traditionally, phenotypic screens were used to test which compounds would induce a desired change in in vitro or in vivo models, focusing on empirical, function-oriented exploration naïve to molecular mechanism (951–953).
In many cases, these screens utilize large libraries that encompass a diverse set of agents varying in many pharmacologically relevant properties (e.g., (954)).
The compounds inducing a desired effect could then be followed up on.
Around the turn of the millennium, advances in molecular biology allowed for HTS to shift towards screening for compounds interacting with a specific molecular target under the hypothesis that modulating that target would have a desired effect.
These approaches both offer pros and cons, and today a popular view is that they are most effective in combination (951, 953, 955).
Today, some efforts to screen compounds for potential repurposing opportunities are experimental, but others use computational HTS approaches (946, 956).
Computational drug repurposing screens can take advantage of big data in biology (737) and as a result are much more feasible today than during the height of the SARS and MERS outbreaks in the early 2000s and early 2010s, respectively.
Advancements in robotics also facilitate the experimental component of HTS (948).
For viral diseases, the goal of drug repurposing is typically to identify existing drugs that have an antiviral effect likely to impede the virus of interest.
While both small molecules and biologics can be candidates for repurposing, the significantly lower price of many small molecule drugs means that they are typically more appealing candidates (957).
Depending on the study design, screens vary in how closely they are tied to a hypothesis.
As with the candidate therapeutics described above, high-throughput experimental or computational screens can proceed based on a hypothesis.
Just as remdesivir was selected as a candidate antiviral because it is a nucleoside analog (958), so too can high-throughput screens select libraries of compounds based on a molecular hypothesis.
Likewise, when the library of drugs is selected without basis in a potential mechanism, a screen can be considered hypothesis free (958).
Today, both types of analyses are common both experimentally and computationally.
Both strategies have been applied to identifying candidate therapeutics against SARS-CoV-2.
4.7.1 Hypothesis-Driven Screening
Hypothesis-driven screens often select drugs likely to interact with specific viral or host targets or drugs with desired clinical effects, such as immunosuppressants.
There are several properties that might identify a compound as a candidate for an emerging viral disease.
Drugs that interact with a target that is shared between pathogens (i.e., a viral protease or a polymerase) or between a viral pathogen and another illness (i.e., a cancer drug with antiviral potential) are potential candidates, as are drugs that are thought to interact with additional molecular targets beyond those they were developed for (956).
Such research can be driven by in vitro or in silico experimentation.
Computational analyses depend on identifying compounds that modulate pre-selected proteins in the virus or host.
As a result, they build on experimental research characterizing the molecular features of the virus, host, and candidate compounds (949).
One example of the application of this approach to COVID-19 research comes from work on protease inhibitors.
Studies have shown that viral proteases play an important role in the life cycle of viruses, including coronaviruses, by modulating the cleavage of viral polyprotein precursors (959).
Several FDA-approved drugs target proteases, such as lopinavir and ritonavir for HIV infection and simeprevir for hepatitis C virus infection.
Serine protease inhibitors were previously suggested as possible treatments for SARS and MERS (960).
One early study (33) suggested that camostat mesylate, a protease inhibitor, could block the entry of SARS-CoV-2 into lung cells in vitro.
Two polyproteins encoded by the SARS-CoV-2 replicase gene, pp1a and pp1ab, are critical for viral replication and transcription (961).
These polyproteins must undergo proteolytic processing, which is usually conducted by main protease (MPro), a 33.8-kDa SARS-CoV-2 protease that is therefore fundamental to viral replication and transcription.
Therefore, it was hypothesized that compounds targeting MPro could be used to prevent or slow the replication of the SARS-CoV-2 virus.
Both computational and experimental approaches facilitated the identification of compounds that might inhibit SARS-CoV-2 MPro.
In 2005, computer-aided design facilitated the development of a Michael acceptor inhibitor, now known as N3, to target MPro of SARS-like coronaviruses (962).
N3 binds in the substrate binding pocket of MPro in several HCoV (962–965).
The structure of N3-bound SARS-CoV-2 MPro has been solved, confirming the computational prediction that N3 would similarly bind in the substrate binding pocket of SARS-CoV-2 (961).
N3 was tested in vitro on SARS-CoV-2-infected Vero cells, which belong to a line of cells established from the kidney epithelial cells of an African green monkey, and was found to inhibit SARS-CoV-2 (961).
A library of approximately 10,000 compounds was screened in a fluorescence resonance energy transfer assay constructed using SARS-CoV-2 MPro expressed in Escherichia coli(961).
Six leads were identified in this hypothesis-driven screen.
In vitro analysis revealed that ebselen had the strongest potency in reducing the viral load in SARS-CoV-2-infected Vero cells (961).
Ebselen is an organoselenium compound with anti-inflammatory and antioxidant properties (966).
Molecular dynamics analysis further demonstrated the potential for ebselen to bind to MPro and disrupt the protease’s enzymatic functions (967).
However, ebselen is likely to be a promiscuous binder, which could diminish its therapeutic potential (961, 968), and compounds with higher specificity may be needed to translate this mechanism effectively to clinical trials.
In July 2020, phase II clinical trials commenced to assess the effects of SPI-1005, an investigational drug from Sound Pharmaceuticals that contains ebselen (969), on 60 adults presenting with each of moderate (970) and severe (971) COVID-19.
Other MPro inhibitors are also being evaluated in clinical trials (972, 973, 973).
Pending the results of clinical trials, N3 remains a computationally interesting compound based on both computational and experimental data, but whether these potential effects will translate to the clinic remains unknown.
4.7.2 Hypothesis-Free Screening
Hypothesis-free screens use a discovery-driven approach, where screens are not targeted to specific viral proteins, host proteins, or desired clinical modulation.
Hypothesis-free drug screening began twenty years ago with the testing of libraries of drugs experimentally.
Today, like many other areas of biology, in silico analyses have become increasingly popular and feasible through advances in biological big data (958, 974).
Many efforts have collected data about interactions between drugs and SARS-CoV-2 and about the host genomic response to SARS-CoV-2 exposure, allowing for hypothesis-free computational screens that seek to identify new candidate therapeutics.
Thus, they utilize a systems biology paradigm to extrapolate the effect of a drug against a virus based on the host interactions with both the virus and the drug (949).
Resources such as the COVID-19 Drug and Gene Set Library, which at the time of its publication contained 1,620 drugs sourced from 173 experimental and computational drug sets and 18,676 human genes sourced from 444 gene sets (975), facilitate such discovery-driven approaches.
Analysis of these databases indicated that some drugs had been identified as candidates across multiple independent analyses, including high-profile candidates such as CQ/HCQ and remdesivir (975).
Computational screening efforts can then mine databases and other resources to identify potential PPIs among the host, the virus, and established and/or experimental drugs (976).
Subject matter expertise from human users may be integrated to varying extents depending on the platform (e.g,. (976, 977)).
These resources have allowed studies to identify potential therapeutics for COVID-19 without an a priori reason for selecting them.
One example of a hypothesis-free screen for COVID-19 drugs comes from a PPI-network-based analysis that was published early in the pandemic (193).
Here, researchers cloned the proteins expressed by SARS-CoV-2 in vitro and quantified 332 viral-host PPI using affinity purification mass spectrometry (193).
They identified two SARS-CoV-2 proteins (Nsp6 and Orf9c) that interacted with host Sigma-1 and Sigma-2 receptors.
Sigma receptors are located in the endoplasmic reticulum of many cell types, and type 1 and 2 Sigma receptors have overlapping but distinct affinities for a variety of ligands (978).
Molecules interacting with the Sigma receptors were then analyzed and found to have an effect on viral infectivity in vitro(193).
A follow-up study evaluated the effect of perturbing these 332 proteins in two cells lines, A549 and Caco-2, using knockdown and knockout methods, respectively, and found that the replication of SARS-CoV-2 in cells from both lines was dependent on the expression of SIGMAR1, which is the gene that encodes the Sigma-1 receptor (979).
Following these results, drugs interacting with Sigma receptors were suggested as candidates for repurposing for COVID-19 (e.g., (980)).
Because many well-known and affordable drugs interact with the Sigma receptors (193, 981), they became a major focus of drug repurposing efforts.
Some of the drugs suggested by the apparent success of Sigma receptor-targeting drugs were already being investigated at the time.
HCQ, for example, forms ligands with both Sigma-1 and Sigma-2 receptors and was already being explored as a candidate therapeutic for COVID-19 (193).
Thus, this computational approach yielded interest in drugs whose antiviral activity was supported by initial in vitro analyses.
Follow-up research, however, called into question whether the emphasis on drugs interacting with Sigma receptors might be based on a spurious association (982).
This study built on the prior work by examining whether antiviral activity among compounds correlated with their affinity for the Sigma receptors and found that it did not.
The study further demonstrated that cationic amphiphilicity was a shared property among many of the candidate drugs identified through both computational and phenotypic screens and that it was likely to be the source of many compounds’ proposed antiviral activity (982).
Cationic amphiphilicity is associated with the induction of phospholipidosis, which is when phospholipids accumulate in the lysosome (983).
Phospholipidosis can disrupt viral replication by inhibiting lipid processing (984) (see discussion of HCQ in Appendix 1).
However, phospholipidosis is known to translate poorly from in vitro models to in vivo models or clinical applications.
Thus, this finding suggested that these screens were identifying compounds that shared a physiochemical property rather than a specific target (982).
The authors further demonstrated that antiviral activity against SARS-CoV-2 in vitro was correlated with the induction of phospholipidosis for drugs both with and without cationic amphiphilicity (982).
This finding supports the idea that the property of cationic amphicility was being detected as a proxy for the shared effect of phospholipidosis (982).
They demonstrated that phospholipidosis-inducing drugs were not effective at preventing viral propagation in vivo in a murine model of COVID-19 (982).
Therefore, removing hits that induce phospholipidosis from computational and in vitro experimental repurposing screens (e.g., (985)) may help emphasize those that are more likely to provide clinical benefits.
This work illustrates the importance of considering confounding variables in computational screens, a principle that has been incorporated into more traditional approaches to drug development (986).
One drug that acts on Sigma receptors does, however, remain a candidate for the treatment of COVID-19.
Several psychotropic drugs target Sigma receptors in the central nervous system and thus attracted interest as potential COVID-19 therapeutics following the findings of two host-virus PPI studies (987).
For several of these drugs, the in vitro antiviral activity (979) was not correlated with their affinity for the Sigma-1 receptor (982, 987) but was correlated with phospholipidosis (982).
However, fluvoxamine, a selective serotonin reuptake inhibitor that is a particularly potent Sigma-1 receptor agonist (987), has shown promise as a preventative of severe COVID-19 in a preliminary analysis of data from the large-scale TOGETHER trial (863).
As of August 6, 2021, this trial had collected data from over 1,400 patients in the fluvoxamine arm of their study, half of whom received a placebo (863).
Only 74 patients in the fluvoxamine group had progressed to hospitalization for COVID-19 compared to 107 in the placebo group, corresponding to a relative risk of 0.69; additionally, the relative risk of mortality between the two groups was calculated at 0.71.
These findings support the results of small clinical trials that have found fluvoxamine to reduce clinical deterioration relative to a placebo (988, 989).
However, the ongoing therapeutic potential of fluvoxamine does not contradict the finding that hypothesis-free screening hits can be driven by confounding factors.
The authors point out that its relevance would not just be antiviral as it has a potential immunomodulatory mechanism (988).
It has been found to be protective against septic shock in an in vivo mouse model (990).
It is possible that fluvoxamine also exerts an antiviral effect (991).
Thus, Sigma-1 receptor activity may contribute to fluvoxamine’s potential effects in treating COVID-19, but is not the only mechanism by which this drug can interfere with disease progression.
4.7.3 Potential and Limitations of High-Throughput Analyses
Computational screening allows for a large number of compounds to be evaluated to identify those most likely to display a desired behavior or function.
This approach can be guided by a hypothesis or can aim to discover underlying characteristics that produce new hypotheses about the relationship between a host, a virus, and candidate pharmaceuticals.
The examples outlined above illustrate that HTS-based evaluations of drug repurposing can potentially provide valuable insights.
Computational techniques were used to design compounds targeting MPro based on an understanding of how this protease aids viral replication, and MPro inhibitors remain promising candidates (948), although the clinical trial data is not yet available.
Similarly, computational analysis correctly identified the Sigma-1 receptor as a protein of interest.
Although the process of identifying which drugs might modulate the interaction led to an emphasis on candidates that ultimately have not been supported, fluvoxamine remains an appealing candidate.
The difference between the preliminary evidence for fluvoxamine compared to other drugs that interact with Sigma receptors underscores a major critique of hypothesis-free HTS in particular: while these approaches allow for brute force comparison of a large number of compounds against a virus of interest, they lose the element of expertise that is associated with most successes in drug repurposing (958).
There are also practical limitations to these methods.
One concern is that computational analyses inherently depend on the quality of the data being evaluated.
The urgency of the COVID-19 pandemic led many research groups to pivot towards computational HTS research without familiarity with best practices in this area (948).
As a result, there is an excessive amount of information available from computational studies (992), but not all of it is high-quality.
Additionally, the literature used to identify and validate targets can be difficult to reproduce (993), which may pose challenges to target-based experimental screening and to in silico screens.
Some efforts to repurpose antivirals have focused on host, rather than viral, proteins (949), which might be expected to translate poorly in vivo if the targeted proteins serve essential functions in the host.
Concerns about the practicality of hypothesis-free screens to gain novel insights are underscored by the fact that very few or possibly no success stories have emerged from hypothesis-free screens over the past twenty years (958).
These findings suggest that data-driven research can be an important component of the drug repurposing ecosystem, but that drug repurposing efforts that proceed without a hypothesis, an emphasis on biological mechanisms, or an understanding of confounding effects may not produce viable candidates.
4.8 Considerations in Balancing Different Approaches
The approaches described here offer a variety of advantages and limitations in responding to a novel viral threat and building on existing bodies of knowledge in different ways.
Medicine, pharmacology, basic science (especially virology and immunology), and biological data science can all provide different insights and perspectives for addressing the challenging question of which existing drugs might provide benefits against an emerging viral threat.
A symptom management-driven approach allows clinicians to apply experience with related diseases or related symptoms to organize a rapid response aimed at saving the lives of patients already infected with a new disease.
Oftentimes, the pharmaceutical agents that are applied are small-molecule, broad-spectrum pharmaceuticals that are widely available and affordable to produce, and they may already be available for other purposes, allowing clinicians to administer them to patients quickly either with an EUA or off-label.
In this vein, dexamethasone has emerged as the strongest treatment against severe COVID-19 (Table 1).
Alternatively, many efforts to repurpose drugs for COVID-19 have built on information gained through basic scientific research of HCoV.
Understanding how related viruses function has allowed researchers to identify possible pharmacological strategies to disrupt pathogenesis (Figure 5).
Some of the compounds identified through these methods include small-molecule antivirals, which can be boutique and experimental medications like remdesivir (Table 1).
Other candidate drugs that intercept host-pathogen interactions include biologics, which imitate the function of endogenous host compounds.
Most notably, several mAbs that have been developed (casirivimab, imdevimab, bamlanivimab and etesevimab) or repurposed (sotrovimab, tocilizumab) have now been granted EUAs (Table 1).
Although not discussed here, several vaccine development programs have also met huge success using a range of strategies (5, 6).
Table 1: Summary table of candidate therapeutics examined in this manuscript.
“Grade” is the rating given to each treatment by the Systematic Tracker of Off-label/Repurposed Medicines Grades (STORM) maintained by the Center for Cytokine Storm Treatment & Laboratory (CSTL) at the University of Pennsylvania (788).
A grade of A indicates that a treatment is considered effective, B that all or most RCTs have shown positive results, C that RCT data are not yet available, and D that multiple RCTs have produced negative results.
Treatments not in the STORM database are indicated as N/A.
FDA status is also provided where available.
The evidence available is based on the progression of the therapeutic through the pharmaceutical development pipeline, with RCTs as the most informative source of evidence.
The effectiveness is summarized based on the current available evidence; large trials such as RECOVERY and Solidarity are weighted heavily in this summary.
This table was last updated on August 20, 2021.
Treatment
Grade
Category
FDA Status
Evidence Available
Suggested Effectiveness
Dexamethasone
A
Small molecule, broad spectrum
Used off-label
RCT
Supported: RCT shows improved outcomes over SOC, especially in severe cases such as CRS
Remdesivir
A
Small molecule, antiviral, adenosine analog
Approved for COVID-19 (and EUA for combination with baricitinib)
RCT
Mixed: Conflicting evidence from large WHO-led Solidarity trial vs US-focused RCT and other studies
Tocilizumab
A
Biologic, monoclonal antibody
EUA
RCT
Mixed: It appears that TCZ may work well in combination with dexamethasone in severe cases, but not as monotherapy
Supported: Phase 2 clinical trial showed reduction in viral load, but FDA pause recommended because may be less effective against Delta variant
Casirivimab and imdevimab
N/A
Biologic, monoclonal antibodies
EUA
RCT
Supported: Reduced viral load at interim analysis
Fluvoxamine
B
Small-molecule, Sigma-1 receptor agonist
N/A
RCT
Supported: Support from two small RCTs and preliminary support from interim analysis of TOGETHER
SNG001
B
Biologic, interferon
None
RCT
Mixed: Support from initial RCT but no effect found in WHO’s Solidarity trial
MPro Protease Inhibitors
N/A
Small molecule, protease inhibitor
None
Computational prediction, in vitro studies
Unknown
ARBs & ACEIs
C
Small molecule, broad spectrum
None
Observational studies and some RCTs
Not supported: Observational study retracted, RCTs suggest no association
Favipiravir
D
Small molecule, antiviral, nucleoside analog
None
RCT
Not supported: RCTs do not show significant improvements for individuals taking this treatment, good safety profile
HCQ/CQ
D
Small molecule, broad spectrum
None
RCT
Not supported, possibly harmful: Non-blinded RCTs showed no improvement over SOC, safety profile may be problematic
Convalescent plasma transfusion
D
Biologic, polyclonal antibodies
EUA
RCT
Mixed: Supported in small trials but not in large-scale RECOVERY trial
Ivermectin
D
Small molecule, broad spectrum
None
RCT
Mixed: Mixed results from small RCTs, major supporting RCT now withdrawn, preliminary results of large RCT (TOGETHER) suggest no effect on emergency room visits or hospitalization for COVID-19
All of the small-molecule drugs evaluated and most of the biologics are repurposed, and thus hinge on a theoretical understanding of how the virus interacts with a human host and how pharmaceuticals can be used to modify those interactions rather than being designed specifically against SARS-CoV-2 or COVID-19.
As a result, significant attention has been paid to computational approaches that automate the identification of potentially desirable interactions.
However, work in COVID-19 has made it clear that relevant compounds can also be masked by confounds, and spurious associations can drive investment in candidate therapeutics that are unlikely to translate to the clinic.
Such spurious hits are especially likely to impact hypothesis-free screens.
However, hypothesis-free screens may still be able to contribute to the drug discovery or repurposing ecosystem, assuming the computational arm of HTS follows the same trends seen in its experimental arm.
In 2011, a landmark study in drug discovery demonstrated that although more new drugs were discovered using target-based rather than phenotypic approaches, the majority of drugs with a novel molecular mechanism of action (MMOA) were identified in phenotypic screens (994).
This pattern applied only to first-in-class drugs, with most follower drugs produced by target-based screening (952).
These findings suggest that target-based drug discovery is more successful when building on a known MMOA, and that modulating a target is most valuable when the target is part of a valuable MMOA (953).
Building on this, many within the field suggested that mechanism-informed phenotypic investigations may be the most useful approach to drug discovery (951, 953, 955).
As it stands, data-driven efforts to identify patterns in the results of computational screens allowed researchers to notice the shared property of cationic amphicility among many of the hits from computational screening analyses (982).
While easier said than done, efforts to fill in the black box underlying computational HTS and recognize patterns among the identified compounds aid in moving data-oriented drug repurposing efforts in this direction.
The unpredictable nature of success and failure in drug repurposing for COVID-19 thus highlights one of the tenets of phenotypic screening: there are a lot of “unknown unknowns”, and a promising mechanism at the level of an MMOA will not necessarily propagate up to the pathway, cellular, or organismal level (951).
Despite the fact that apparently mechanistically relevant drugs may exist, identifying effective treatments for a new viral disease is extremely challenging.
Targets of repurposed drugs are often non-specific, meaning that the MMOA can appear to be relevant to COVID-19 without a therapeutic or prophylactic effect being observed in clinical trials.
The difference in the current status of remdesivir and favipiravir as treatments for COVID-19 (Table 1) underscores how difficult to predict whether a specific compound will produce a desired effect, even when the mechanisms are similar.
Furthermore, the fact that many candidate COVID-19 therapeutics were ultimately identified because of their shared propensity to induce phospholipidosis underscores how challenging it can be to identify a mechanism in silico or in vitro that will translate to a successful treatment.
While significant progress has been made thus far in the pandemic, the therapeutic landscape is likely to continue to evolve as more results become available from clinical trials and as efforts to develop novel therapeutics for COVID-19 progress.
4.9 Towards the Next HCoV Threat
Only very limited testing of candidate therapies was feasible during the SARS and MERS epidemics, and as a result, few treatments were available at the outset of the COVID-19 pandemic.
Even corticosteroids, which were used to treat SARS patients, were a controversial therapeutic prior to the release of the results of the large RECOVERY trial.
The scale and duration of the COVID-19 pandemic has made it possible to conduct large, rigorous RCTs such as RECOVERY, Solidarity, TOGETHER, and others.
As results from these trials have continued to emerge, it has become clear that small clinical trials often produce spurious results.
In the case of HCQ/CQ, the therapeutic had already attracted so much attention based on small, preliminary (and in some cases, methodologically concerning) studies that it took the results of multiple large studies before attention began to be redirected to more promising candidates (995).
In fact, most COVID-19 clinical trials lack the statistical power to reliably test their hypotheses (996, 997).
In the face of an urgent crisis like COVID-19, the desire to act quickly is understandable, but it is imperative that studies maintain strict standards of scientific rigor (948, 986), especially given the potential dangers of politicization, as illustrated by HCQ/CQ (998).
Potential innovations in clinical trial structure, such as adaptable clinical trials with master protocols (999) or the sharing of data among small clinical trials (997) may help to address future crises and to bolster the results from smaller studies, respectively.
In the long-term, new drugs specific for treatment of COVID-19 may also enter development.
Development of novel drugs is likely to be guided by what is known about the pathogenesis and molecular structure of SARS-CoV-2.
For example, understanding the various structural components of SARS-CoV-2 may allow for the development of small molecule inhibitors of those components.
Crystal structures of the SARS-CoV-2 main protease have been resolved (961, 1000).
Much work remains to be done to determine further crystal structures of other viral components, understand the relative utility of targeting different viral components, perform additional small molecule inhibitor screens, and determine the safety and efficacy of the potential inhibitors.
While still nascent, work in this area is promising.
Over the longer term, this approach and others may lead to the development of novel therapeutics specifically for COVID-19 and SARS-CoV-2.
Such efforts are likely to prove valuable in managing future emergent HCoV, just as research from the SARS and MERS pandemic has provided a basis for the COVID-19 response.
5 Appendix: Identification and Development of Therapeutics for COVID-19
5.1 Dexamethasone
In order to understand how dexamethasone reduces inflammation, it is necessary to consider the stress response broadly.
In response to stress, corticotropin‐releasing hormone stimulates the release of neurotransmitters known as catecholamines, such as epinephrine, and steroid hormones known as glucocorticoids, such as cortisol (1001, 1002).
While catecholamines are often associated with the fight-or-flight response, the specific role that glucocorticoids play is less clear, although they are thought to be important to restoring homeostasis (1003).
Immune challenge is a stressor that is known to interact closely with the stress response.
The immune system can therefore interact with the central nervous system; for example, macrophages can both respond to and produce catecholamines (1001).
Additionally, the production of both catecholamines and glucocorticoids is associated with inhibition of proinflammatory cytokines such as IL-6, IL-12, and tumor necrosis factor-α (TNF‐α) and the stimulation of anti-inflammatory cytokines such as IL-10, meaning that the stress response can regulate inflammatory immune activity (1002).
Administration of dexamethasone has been found to correspond to dose-dependent inhibition of IL-12 production, but not to affect IL-10 (1004); the fact that this relationship could be disrupted by administration of a glucocorticoid-receptor antagonist suggests that it is regulated by the receptor itself (1004).
Thus, the administration of dexamethasone for COVID-19 is likely to simulate the release of glucocorticoids endogenously during stress, resulting in binding of the synthetic steroid to the glucocorticoid receptor and the associated inhibition of the production of proinflammatory cytokines.
In this model, dexamethasone reduces inflammation by stimulating the biological mechanism that reduces inflammation following a threat such as immune challenge.
Initial support for dexamethasone as a treatment for COVID-19 came from the United Kingdom’s RECOVERY trial (780), which assigned over 6,000 hospitalized COVID-19 patients to the standard of care (SOC) or treatment (dexamethasone) arms of the trial at a 2:1 ratio.
At the time of randomization, some patients were ventilated (16%), others were on non-invasive oxygen (60%), and others were breathing independently (24%).
Patients in the treatment arm were administered dexamethasone either orally or intravenously at 6 mg per day for up to 10 days.
The primary end-point was the patient’s status at 28-days post-randomization (mortality, discharge, or continued hospitalization), and secondary outcomes analyzed included the progression to invasive mechanical ventilation over the same period.
The 28-day mortality rate was found to be lower in the treatment group than in the SOC group (21.6% vs 24.6%, p < 0.001).
However, the effect was driven by improvements in patients receiving mechanical ventilation or supplementary oxygen.
One possible confounder is that patients receiving mechanical ventilation tended to be younger than patients who were not receiving respiratory support (by 10 years on average) and to have had symptoms for a longer period.
However, adjusting for age did not change the conclusions, although the duration of symptoms was found to be significantly associated with the effect of dexamethasone administration.
Thus, this large, randomized, and multi-site, albeit not placebo-controlled, study suggests that administration of dexamethasone to patients who are unable to breathe independently may significantly improve survival outcomes.
Additionally, dexamethasone is a widely available and affordable medication, raising the hope that it could be made available to COVID-19 patients globally.
It is not surprising that administration of an immunosuppressant would be most beneficial in severe cases where the immune system was dysregulated towards inflammation.
However, it is also unsurprising that care must be taken in administering an immunosuppressant to patients fighting a viral infection.
In particular, the concern has been raised that treatment with dexamethasone might increase patient susceptibility to concurrent (e.g., nosocomial) infections (1005).
Additionally, the drug could potentially slow viral clearance and inhibit patients’ ability to develop antibodies to SARS-CoV-2 (777, 1005), with the lack of data about viral clearance being put forward as a major limitation of the RECOVERY trial (1006).
Furthermore, dexamethasone has been associated with side effects that include psychosis, glucocorticoid-induced diabetes, and avascular necrosis (777), and the RECOVERY trial did not report outcomes with enough detail to be able to determine whether they observed similar complications.
The effects of dexamethasone have also been found to differ among populations, especially in high-income versus middle- or low-income countries (1007).
However, since the RECOVERY trial’s results were released, strategies have been proposed for administering dexamethasone alongside more targeted treatments to minimize the likelihood of negative side effects (1005).
Given the available evidence, dexamethasone is currently the most promising treatment for severe COVID-19.
5.2 Favipiravir
The effectiveness of favipiravir for treating patients with COVID-19 is currently under investigation.
Evidence for the drug inhibiting viral RNA polymerase are based on time-of-drug addition studies that found that viral loads were reduced with the addition of favipiravir in early times post-infection (798, 801, 802).
An open-label, nonrandomized, before-after controlled study for COVID-19 was recently conducted (1008).
The study included 80 COVID-19 patients (35 treated with favipiravir, 45 control) from the isolation ward of the National Clinical Research Center for Infectious Diseases (The Third People’s Hospital of Shenzhen), Shenzhen, China.
The patients in the control group were treated with other antivirals, such as lopinavir and ritonavir.
It should be noted that although the control patients received antivirals, two subsequent large-scale analyses, the WHO Solidarity trial and the Randomized Evaluation of COVID-19 Therapy (RECOVERY) trial, identified no effect of lopinavir or of a lopinavir-ritonavir combination, respectively, on the metrics of COVID-19-related mortality that each assessed (811, 1009, 1010).
Treatment was applied on days 2-14; treatment stopped either when viral clearance was confirmed or at day 14.
The efficacy of the treatment was measured by, first, the time until viral clearance using Kaplan-Meier survival curves, and, second, the improvement rate of chest computed tomography (CT) scans on day 14 after treatment.
The study found that favipiravir increased the speed of recovery, measured as viral clearance from the patient by RT-PCR, with patients receiving favipiravir recovering in four days compared to 11 days for patients receiving antivirals such as lopinavir and ritonavir.
Additionally, the lung CT scans of patients treated with favipiravir showed significantly higher improvement rates (91%) on day 14 compared to control patients (62%, p = 0.004).
However, there were adverse side effects in 4 (11%) favipiravir-treated patients and 25 (56%) control patients.
The adverse side effects included diarrhea, vomiting, nausea, rash, and liver and kidney injury.
Despite the study reporting clinical improvement in favipiravir-treated patients, several study design issues are problematic and lower confidence in the overall conclusions.
For example, the study was neither randomized nor blinded.
Moreover, the selection of patients did not take into consideration important factors such as previous clinical conditions or sex, and there was no age categorization.
Additionally, it should be noted that this study was temporarily retracted and then restored without an explanation (1011).
In late 2020 and early 2021, the first randomized controlled trials of favipiravir for the treatment of COVID-19 released results (1012–1014).
One study (1013) was retracted in November 2021 due to concerns about the data.
Of the two remaining, the first (1012) used a randomized, controlled, open-label design to compare two drugs, favipiravir and baloxavir marboxil, to SOC alone.
Here, SOC included antivirals such as lopinavir/ritonavir and was administered to all patients.
The primary endpoint analyzed was viral clearance at day 14.
The sample size for this study was very small, with 29 total patients enrolled, and no significant effect of the treatments was found for the primary or any of the secondary outcomes analyzed, which included mortality.
The second trial examined 60 patients and reported a significant effect of favipiravir on viral clearance at four days (a secondary endpoint), but not at 10 days (the primary endpoint) (1014).
This study, as well as a prior study of favipiravir (1015), also reported that the drug was generally well-tolerated.
Thus, in combination, these small studies suggest that the effects of favipiravir as a treatment for COVID-19 cannot be determined based on the available evidence, but additionally, none raise major concerns about the safety profile of the drug.
5.3 Remdesivir
At the outset of the COVID-19 pandemic, remdesivir did not have any have any FDA-approved use.
A clinical trial in the Democratic Republic of Congo found some evidence of effectiveness against ebola virus disease (EVD), but two antibody preparations were found to be more effective, and remdesivir was not pursued (1016).
Remdesivir also inhibits polymerase and replication of the coronaviruses MERS-CoV and SARS-CoV-1 in cell culture assays with submicromolar IC50s (1017).
It has also been found to inhibit SARS-CoV-2, showing synergy with CQ in vitro(806).
Remdesivir was first used on some COVID-19 patients under compassionate use guidelines (1020).
All were in late stages of COVID-19 infection, and initial reports were inconclusive about the drug’s efficacy.
Gilead Sciences, the maker of remdesivir, led a recent publication that reported outcomes for compassionate use of the drug in 61 patients hospitalized with confirmed COVID-19.
Here, 200 mg of remdesivir was administered intravenously on day 1, followed by a further 100 mg/day for 9 days (810).
There were significant issues with the study design, or lack thereof.
There was no randomized control group.
The inclusion criteria were variable: some patients only required low doses of oxygen, while others required ventilation.
The study included many sites, potentially with variable inclusion criteria and treatment protocols.
The patients analyzed had mixed demographics.
There was a short follow-up period of investigation.
Eight patients were excluded from the analysis mainly due to missing post-baseline information; thus, their health was unaccounted for.
Therefore, even though the study reported clinical improvement in 68% of the 53 patients ultimately evaluated, due to the significant issues with study design, it could not be determined whether treatment with remdesivir had an effect or whether these patients would have recovered regardless of treatment.
Another study comparing 5- and 10-day treatment regimens reported similar results but was also limited because of the lack of a placebo control (1021).
These studies did not alter the understanding of the efficacy of remdesivir in treating COVID-19, but the encouraging results provided motivation for placebo-controlled studies.
The double-blind placebo-controlled ACTT-1 trial (807, 808) recruited 1,062 patients and randomly assigned them to placebo treatment or treatment with remdesivir.
Patients were stratified for randomization based on site and the severity of disease presentation at baseline (807).
The treatment was 200 mg on day 1, followed by 100 mg on days 2 through 10.
Data was analyzed from a total of 1,059 patients who completed the 29-day course of the trial, with 517 assigned to remdesivir and 508 to placebo (807).
The two groups were well matched demographically and clinically at baseline.
Those who received remdesivir had a median recovery time of 10 days, as compared with 15 days in those who received placebo (rate ratio for recovery, 1.29; 95% confidence interval (CI), 1.12 to 1.49; p < 0.001).
The Kaplan-Meier estimates of mortality by 14 days were 6.7% with remdesivir and 11.9% with placebo, with a hazard ratio (HR) for death of 0.55 and a 95% CI of 0.36 to 0.83, and at day 29, remdesivir corresponded to 11.4% and the placebo to 15.2% (HR: 0.73; 95% CI, 0.52 to 1.03).
Serious adverse events were reported in 131 of the 532 patients who received remdesivir (24.6%) and in 163 of the 516 patients in the placebo group (31.6%).
This study also reported an association between remdesivir administration and both clinical improvement and a lack of progression to more invasive respiratory intervention in patients receiving non-invasive and invasive ventilation at randomization (807).
Largely on the results of this trial, the FDA reissued and expanded the EUA for remdesivir for the treatment of hospitalized COVID-19 patients ages twelve and older (1022).
Additional clinical trials (806, 1023–1026) are currently underway to evaluate the use of remdesivir to treat COVID-19 patients at both early and late stages of infection and in combination with other drugs (Figure 4).
As of October 22, 2020, remdesivir received FDA approval based on three clinical trials (1027).
However, results suggesting no effect of remdesivir on survival were reported by the WHO Solidarity trial (811).
Patients were randomized in equal proportions into four experimental conditions and a control condition, corresponding to four candidate treatments for COVID-19 and SOC, respectively; no placebo was administered.
The 2,750 patients in the remdesivir group were administered 200 mg intravenously on the first day and 100 mg on each subsequent day until day 10 and assessed for in-hospital death (primary endpoint), duration of hospitalization, and progression to mechanical ventilation.
There were also 2,708 control patients who would have been eligible and able to receive remdesivir were they not assigned to the control group.
A total of 604 patients among these two cohorts died during initial hospitalization, with 301 in the remdesivir group and 303 in the control group.
The rate ratio of death between these two groups was therefore not significant (0.95, p = 0.50), suggesting that the administration of remdesivir did not affect survival.
The two secondary analyses similarly did not find any effect of remdesivir.
Additionally, the authors compared data from their study with data from three other studies of remdesivir (including (807)) stratified by supplemental oxygen status.
A meta-analysis of the four studies yielded an overall rate ratio for death of 0.91 (p = 0.20).
These results thus do not support the previous findings that remdesivir reduced median recovery time and mortality risk in COVID-19 patients.
In response to the results of the Solidarity trial, Gilead, which manufactures remdesivir, released a statement pointing to the fact that the Solidarity trial was not placebo-controlled or double-blind and at the time of release, the statement had not been peer reviewed (1028); these sentiments have been echoed elsewhere (1029).
Other critiques of this study have noted that antivirals are not typically targeted at patients with severe illness, and therefore remdesivir could be more beneficial for patients with mild rather than severe cases (1010, 1030).
However, the publication associated with the trial sponsored by Gilead did purport an effect of remdesivir on patients with severe disease, identifying an 11 versus 18 day recovery period (rate ratio for recovery: 1.31, 95% CI 1.12 to 1.52) (807).
Additionally, a smaller analysis of 598 patients, of whom two-thirds were randomized to receive remdesivir for either 5 or 10 days, reported a small effect of treatment with remdesivir for five days relative to standard of care in patients with moderate COVID-19 (1031).
These results suggest that remdesivir could improve outcomes for patients with moderate COVID-19, but that additional information would be needed to understand the effects of different durations of treatment.
Therefore, the Solidarity trial may point to limitations in the generalizability of other research on remdesivir, especially since the broad international nature of the Solidarity clinical trial, which included countries with a wide range of economic profiles and a variety of healthcare systems, provides a much-needed global perspective in a pandemic (1010).
On the other hand, only 62% of patients in the Solidarity trial were randomized on the day of admission or one day afterwards (811), and concerns have been raised that differences in disease progression could influence the effectiveness of remdesivir (1010).
Despite the findings of the Solidarity trial, remdesivir remains available for the treatment of COVID-19 in many places.
Remdesivir has also been investigated in combination with other drugs, such as baricitinib, which is an inhibitor of Janus kinase 1 and 2 (1032); the FDA has issued an EUA for the combination of remdesivir and baricitinib in adult and pediatric patients (1033).
Follow-up studies are needed and, in many cases, are underway to further investigate remdesivir-related outcomes.
Similarly, the extent to which the remdesivir dosing regimen could influence outcomes continues to be under consideration.
A randomized, open-label trial compared the effect of remdesivir on 397 patients with severe COVID-19 over 5 versus 10 days (809, 1021), complementing the study that found that a 5-day course of remdesivir improved outcomes for patients with moderate COVID-19 but a 10-day course did not (1031).
Patients in the two groups were administered 200 mg of remdesivir intravenously on the first day, followed by 100 mg on the subsequent four or nine days, respectively.
The two groups differed significantly in their clinical status, with patients assigned to the 10-day group having more severe illness.
This study also differed from most because it included not only adults, but also pediatric patients as young as 12 years old.
It reported no significant differences across several outcomes for patients receiving a 5-day or 10-day course, when correcting for baseline clinical status.
The data did suggest that the 10-day course might reduce mortality in the most severe patients at day 14, but the representation of this group in the study population was too low to justify any conclusions (1021).
Thus, additional research is also required to determine whether the dosage and duration of remdesivir administration influences outcomes.
In summary, remdesivir is the first FDA approved anti-viral against SARS-CoV-2 as well as the first FDA approved COVID-19 treatment.
Early investigations of this drug established proof of principle that drugs targeting the virus can benefit COVID-19 patients.
Moreover, one of the most successful strategies for developing therapeutics for viral diseases is to target the viral replication machinery, which are typically virally encoded polymerases.
Small molecule drugs targeting viral polymerases are the backbones of treatments for other viral diseases including human immunodeficiency virus (HIV) and herpes.
Notably, the HIV and herpes polymerases are a reverse transcriptase and a DNA polymerase, respectively, whereas SARS-CoV-2 encodes an RdRP, so most of the commonly used polymerase inhibitors are not likely to be active against SARS-CoV-2.
In clinical use, polymerase inhibitors show short term benefits for HIV patients, but for long term benefits they must be part of combination regimens.
They are typically combined with protease inhibitors, integrase inhibitors, and even other polymerase inhibitors.
Remdesivir provides evidence that a related approach may be beneficial for the treatment of COVID-19.
5.4 Hydroxychloroquine and Chloroquine
CQ and hydroxychloroquine (HCQ) increase cellular pH by accumulating in their protonated form inside lysosomes (813, 1034).
This shift in pH inhibits the breakdown of proteins and peptides by the lysosomes during the process of proteolysis (813).
Interest in CQ and HCQ for treating COVID-19 was catalyzed by a mechanism observed in in vitro studies of both SARS-CoV-1 and SARS-CoV-2.
In one study, CQ inhibited viral entry of SARS-CoV-1 into Vero E6 cells, a cell line that was derived from Vero cells in 1968, through the elevation of endosomal pH and the terminal glycosylation of ACE2 (814).
Increased pH within the cell, as discussed above, inhibits proteolysis, and terminal glycosylation of ACE2 is thought to interfere with virus-receptor binding.
An in vitro study of SARS-CoV-2 infection of Vero cells found both HCQ and CQ to be effective in inhibiting viral replication, with HCQ being more potent (815).
Additionally, an early case study of three COVID-19 patients reported the presence of antiphospholipid antibodies in all three patients (100).
Antiphospholipid antibodies are central to the diagnosis of the antiphospholipid syndrome, a disorder that HCQ has often been used to treat (1035–1037).
Because the 90% effective concentration (EC90) of CQ in Vero E6 cells (6.90 μM) can be achieved in and tolerated by rheumatoid arthritis (RA) patients, it was hypothesized that it might also be possible to achieve the effective concentration in COVID-19 patients (1038).
Additionally, clinical trials have reported HCQ to be effective in treating HIV (1039) and chronic Hepatitis C (1040).
Together, these studies triggered initial enthusiasm about the therapeutic potential for HCQ and CQ against COVID-19.
HCQ/CQ has been proposed both as a treatment for COVID-19 and a prophylaxis against SARS-CoV-2 exposure, and trials often investigated these drugs in combination with azithromycin (AZ) and/or zinc supplementation.
However, as more evidence has emerged, it has become clear that HCQ/CQ offer no benefits against SARS-CoV-2 or COVID-19.
5.4.1 Trials Assessing Therapeutic Administration of HCQ/CQ
The initial study evaluating HCQ as a treatment for COVID-19 patients was published on March 20, 2020 by Gautret et al. (816).
This non-randomized, non-blinded, non-placebo clinical trial compared HCQ to SOC in 42 hospitalized patients in southern France.
It reported that patients who received HCQ showed higher rates of virological clearance by nasopharyngeal swab on days 3-6 when compared to SOC.
This study also treated six patients with both HCQ + AZ and found this combination therapy to be more effective than HCQ alone.
However, the design and analyses used showed weaknesses that severely limit interpretability of results, including the small sample size and the lack of: randomization, blinding, placebo (no “placebo pill” given to SOC group), Intention-To-Treat analysis, correction for sequential multiple comparisons, and trial pre-registration.
Furthermore, the trial arms were entirely confounded by the hospital and there were false negative outcome measurements (see (1041)).
Two of these weaknesses are due to inappropriate data analysis and can therefore be corrected post hoc by recalculating the p-values (lack of Intention-To-Treat analysis and multiple comparisons).
However, all other weaknesses are fundamental design flaws and cannot be corrected for.
Thus, the conclusions cannot be generalized outside of the study.
The International Society of Antimicrobial Chemotherapy, the scientific organization that publishes the journal where the article appeared, subsequently announced that the article did not meet its expected standard for publications (817), although it has not been officially retracted.
Because of the preliminary data presented in this study, HCQ treatment was subsequently explored by other researchers.
About one week later, a follow-up case study reported that 11 consecutive patients were treated with HCQ + AZ using the same dosing regimen (1042).
One patient died, two were transferred to the intensive care unit (ICU), and one developed a prolonged QT interval, leading to discontinuation of HCQ + AZ administration.
As in the Gautret et al. study, the outcome assessed was virological clearance at day 6 post-treatment, as measured from nasopharyngeal swabs.
Of the ten living patients on day 6, eight remained positive for SARS-CoV-2 RNA.
Like in the original study, interpretability was severely limited by the lack of a comparison group and the small sample size.
However, these results stand in contrast to the claims by Gautret et al. that all six patients treated with HCQ + AZ tested negative for SARS-CoV-2 RNA by day 6 post-treatment.
This case study illustrated the need for further investigation using robust study design to evaluate the efficacy of HCQ and/or CQ.
On April 10, 2020, a randomized, non-placebo trial of 62 COVID-19 patients at the Renmin Hospital of Wuhan University was released (1043).
This study investigated whether HCQ decreased time to fever break or time to cough relief when compared to SOC (1043).
This trial found HCQ decreased both average time to fever break and average time to cough relief, defined as mild or no cough.
While this study improved on some of the methodological flaws in Gautret et al. by randomizing patients, it also had several flaws in trial design and data analysis that prevent generalization of the results.
These weaknesses include the lack of placebo, lack of correction for multiple primary outcomes, inappropriate choice of outcomes, lack of sufficient detail to understand analysis, drastic disparities between pre-registration (1044) and published protocol (including differences in the inclusion and exclusion criteria, the number of experimental groups, the number of patients enrolled, and the outcome analyzed), and small sample size.
The choice of outcomes may be inappropriate as both fevers and cough may break periodically without resolution of illness.
Additionally, for these outcomes, the authors reported that 23 of 62 patients did not have a fever and 25 of 62 patients did not have a cough at the start of the study, but the authors failed to describe how these patients were included in a study assessing time to fever break and time to cough relief.
It is important to note here that the authors claimed “neither the research performers nor the patients were aware of the treatment assignments.”
This blinding seems impossible in a non-placebo trial because at the very least, providers would know whether they were administering a medication or not, and this knowledge could lead to systematic differences in the administration of care.
Correction for multiple primary outcomes can be adjusted post hoc by recalculating p-values, but all of the other issues were design and statistical weaknesses that cannot be corrected for.
Additionally, disparities between the pre-registered and published protocols raise concerns about experimental design.
The design limitations mean that the conclusions cannot be generalized outside of the study.
A second randomized trial, conducted by the Shanghai Public Health Clinical Center, analyzed whether HCQ increased rates of virological clearance at day 7 in respiratory pharyngeal swabs compared to SOC (1045).
This trial was published in Chinese along with an abstract in English, and only the English abstract was read and interpreted for this review.
The trial found comparable outcomes in virological clearance rate, time to virological clearance, and time to body temperature normalization between the treatment and control groups.
The small sample size is one weakness, with only 30 patients enrolled and 15 in each arm.
This problem suggests the study is underpowered to detect potentially useful differences and precludes interpretation of results.
Additionally, because only the abstract could be read, other design and analysis issues could be present.
Thus, though these studies added randomization to their assessment of HCQ, their conclusions should be interpreted very cautiously.
These two studies assessed different outcomes and reached differing conclusions about the efficacy of HCQ for treating COVID-19; the designs of both studies, especially with respect to sample size, meant that no general conclusions can be made about the efficacy of the drug.
Several widely reported studies on HCQ also have issues with data integrity and/or provenance.
A Letter to the Editor published in BioScience Trends on March 16, 2020 claimed that numerous clinical trials have shown that HCQ is superior to control treatment in inhibiting the exacerbation of COVID-19 pneumonia (1046).
This letter has been cited by numerous primary literature, review articles, and media alike (1047, 1048).
However, the letter referred to 15 pre-registration identifiers from the Chinese Clinical Trial Registry.
When these identifiers are followed back to the registry, most trials claim they are not yet recruiting patients or are currently recruiting patients.
For all of these 15 identifiers, no data uploads or links to publications could be located on the pre-registrations.
At the very least, the lack of availability of the primary data means the claim that HCQ is efficacious against COVID-19 pneumonia cannot be verified.
Similarly, a recent multinational registry analysis (1049) analyzed the efficacy of CQ and HCQ with and without a macrolide, which is a class of antibiotics that includes Azithromycin, for the treatment of COVID-19.
The study observed 96,032 patients split into a control and four treatment conditions (CQ with and without a macrolide; HCQ with and without a macrolide).
They concluded that treatment with CQ or HCQ was associated with increased risk of de novo ventricular arrhythmia during hospitalization.
However, this study has since been retracted by The Lancet due to an inability to validate the data used (1050).
These studies demonstrate that increased skepticism in evaluation of the HCQ/CQ and COVID-19 literature may be warranted, possibly because of the significant attention HCQ and CQ have received as possible treatments for COVID-19 and the politicization of these drugs.
Despite the fact that the study suggesting that CQ/HCQ increased risk of ventricular arrhythmia in COVID-19 patients has now been retracted, previous studies have identified risks associated with HCQ/CQ.
A patient with systemic lupus erythematosus developed a prolonged QT interval that was likely exacerbated by use of HCQ in combination with renal failure (1051).
A prolonged QT interval is associated with ventricular arrhythmia (1052).
Furthermore, a separate study (1053) investigated the safety associated with the use of HCQ with and without macrolides between 2000 and 2020.
The study involved 900,000 cases treated with HCQ and 300,000 cases treated with HCQ + AZ.
The results indicated that short-term use of HCQ was not associated with additional risk, but that HCQ + AZ was associated with an enhanced risk of cardiovascular complications (such as a 15% increased risk of chest pain, calibrated HR = 1.15, 95% CI, 1.05 to 1.26) and a two-fold increased 30-day risk of cardiovascular mortality (calibrated HR = 2.19; 95% CI, 1.22 to 3.94).
Therefore, whether studies utilize HCQ alone or HCQ in combination with a macrolide may be an important consideration in assessing risk.
As results from initial investigations of these drug combinations have emerged, concerns about the efficacy and risks of treating COVID-19 with HCQ and CQ have led to the removal of CQ/HCQ from SOC practices in several countries (1054, 1055).
As of May 25, 2020, WHO had suspended administration of HCQ as part of the worldwide Solidarity Trial (1056), and later the final results of this large-scale trial that compared 947 patients administered HCQ to 906 controls revealed no effect on the primary outcome, mortality during hospitalization (rate ratio: 1.19; p = 0.23)
Additional research has emerged largely identifying HCQ/CQ to be ineffective against COVID-19 while simultaneously revealing a number of significant side effects.
A randomized, open-label, non-placebo trial of 150 COVID-19 patients was conducted in parallel at 16 government-designated COVID-19 centers in China to assess the safety and efficacy of HCQ (1057).
The trial compared treatment with HCQ in conjunction with SOC to SOC alone in 150 infected patients who were assigned randomly to the two groups (75 per group).
The primary endpoint of the study was the negative conversion rate of SARS-CoV-2 in 28 days, and the investigators found no difference in this parameter between the groups (estimated difference between SOC plus HCQ and SOC 4.1%; 95% CI, –10.3% to 18.5%).
The secondary endpoints were an amelioration of the symptoms of the disease such as axillary temperature ≤36.6°C, SpO2 >94% on room air, and disappearance of symptoms like shortness of breath, cough, and sore throat.
The median time to symptom alleviation was similar across different conditions (19 days in HCQ + SOC versus 21 days in SOC, p = 0.97).
Additionally, 30% of the patients receiving SOC+HCQ reported adverse outcomes compared to 8.8% of patients receiving only SOC, with the most common adverse outcome in the SOC+HCQ group being diarrhea (10% versus 0% in the SOC group, p = 0.004).
However, there are several factors that limit the interpretability of this study.
Most of the enrolled patients had mild-to-moderate symptoms (98%), and the average age was 46.
SOC in this study included the use of antivirals (Lopinavir-Ritonavir, Arbidol, Oseltamivir, Virazole, Entecavir, Ganciclovir, and Interferon alfa), which the authors note could influence the results.
Thus, they note that an ideal SOC would need to exclude the use of antivirals, but that ceasing antiviral treatment raised ethical concerns at the time that the study was conducted.
In this trial, the samples used to test for the presence of the SARS-CoV-2 virus were collected from the upper respiratory tract, and the authors indicated that the use of upper respiratory samples may have introduced false negatives (e.g., (71)).
Another limitation of the study that the authors acknowledge was that the HCQ treatment began, on average, at a 16-day delay from the symptom onset.
The fact that this study was open-label and lacked a placebo limits interpretation, and additional analysis is required to determine whether HCQ reduces inflammatory response.
Therefore, despite some potential areas of investigation identified in post hoc analysis, this study cannot be interpreted as providing support for HCQ as a therapeutic against COVID-19.
This study provided no support for HCQ against COVID-19, as there was no difference between the two groups in either negative seroconversion at 28 days or symptom alleviation, and in fact, more severe adverse outcomes were reported in the group receiving HCQ.
Additional evidence comes from a retrospective analysis (1058) that examined data from 368 COVID-19 patients across all United States Veteran Health Administration medical centers.
The study retrospectively investigated the effect of the administration of HCQ (n=97), HCQ + AZ (n=113), and no HCQ (n=158) on 368 patients.
The primary outcomes assessed were death and the need for mechanical ventilation.
Standard supportive care was rendered to all patients.
Due to the low representation of women (N=17) in the available data, the analysis included only men, and the median age was 65 years.
The rate of death was 27.8% in the HCQ-only treatment group, 22.1% in the HCQ + AZ treatment group, and 14.1% in the no-HCQ group.
These data indicated a statistically significant elevation in the risk of death for the HCQ-only group compared to the no-HCQ group (adjusted HR: 2.61, p = 0.03), but not for the HCQ + AZ group compared to the no-HCQ group (adjusted HR: 1.14; p = 0.72).
Further, the risk of ventilation was similar across all three groups (adjusted HR: 1.43, p = 0.48 (HCQ) and 0.43, p = 0.09 (HCQ + AZ) compared to no HCQ).
The study thus showed evidence of an association between increased mortality and HCQ in this cohort of COVID-19 patients but no change in rates of mechanical ventilation among the treatment conditions.
The study had a few limitations: it was not randomized, and the baseline vital signs, laboratory tests, and prescription drug use were significantly different among the three groups.
All of these factors could potentially influence treatment outcome.
Furthermore, the authors acknowledge that the effect of the drugs might be different in females and pediatric subjects, since these subjects were not part of the study.
The reported result that HCQ + AZ is safer than HCQ contradicts the findings of the previous large-scale analysis of twenty years of records that found HCQ + AZ to be more frequently associated with cardiac arrhythmia than HCQ alone (1053); whether this discrepancy is caused by the pathology of COVID-19, is influenced by age or sex, or is a statistical artifact is not presently known.
Finally, findings from the RECOVERY trial were released on October 8, 2020.
This study used a randomized, open-label design to study the effects of HCQ compared to SOC in 11,197 patients at 176 hospitals in the United Kingdom (818).
Patients were randomized into either the control group or one of the treatment arms, with twice as many patients enrolled in the control group as any treatment group.
Of the patients eligible to receive HCQ, 1,561 were randomized into the HCQ arm, and 3,155 were randomized into the control arm.
The demographics of the HCQ and control groups were similar in terms of average age (65 years), proportion female (approximately 38%), ethnic make-up (73% versus 76% white), and prevalence of pre-existing conditions (56% versus 57% overall).
In the HCQ arm of the study, patients received 800 mg at baseline and again after 6 hours, then 400 mg at 12 hours and every subsequent 12 hours.
The primary outcome analyzed was all-cause mortality, and patient vital statistics were reported by physicians upon discharge or death, or else at 28 days following HCQ administration if they remained hospitalized.
The secondary outcome assessed was the combined risk of progression to invasive mechanical ventilation or death within 28 days.
By the advice of an external data monitoring committee, the HCQ arm of the study was reviewed early, leading to it being closed due a lack of support for HCQ as a treatment for COVID-19.
COVID-19-related mortality was not affected by HCQ in the RECOVERY trial (rate ratio, 1.09; 95% CI, 0.97 to 1.23; p = 0.15), but cardiac events were increased in the HCQ arm (0.4 percentage points), as was the duration of hospitalization (rate ratio for discharge alive within 28 days: 0.90; 95% CI, 0.83 to 0.98) and likelihood of progression to mechanical ventilation or death (risk ratio 1.14; 95% CI, 1.03 to 1.27).
This large-scale study thus builds upon studies in the United States and China to suggest that HCQ is not an effective treatment, and in fact may negatively impact COVID-19 patients due to its side effects.
Therefore, though none of the studies have been blinded, examining them together makes it clear that the available evidence points to significant dangers associated with the administration of HCQ to hospitalized COVID-19 patients, without providing any support for its efficacy.
5.4.2 HCQ for the Treatment of Mild Cases
One additional possible therapeutic application of HCQ considered was the treatment of mild COVID-19 cases in otherwise healthy individuals.
This possibility was assessed in a randomized, open-label, multi-center analysis conducted in Catalonia (Spain) (1059).
This analysis enrolled adults 18 and older who had been experiencing mild symptoms of COVID-19 for fewer than five days.
Participants were randomized into an HCQ arm (N=136) and a control arm (N=157), and those in the treatment arm were administered 800 mg of HCQ on the first day of treatment followed by 400 mg on each of the subsequent six days.
The primary outcome assessed was viral clearance at days 3 and 7 following the onset of treatment, and secondary outcomes were clinical progression and time to complete resolution of symptoms.
No significant differences between the two groups were found: the difference in viral load between the HCQ and control groups was 0.01 (95% CI, -0.28 to 0.29) at day 3 and -0.07 (95% CI -0.44 to 0.29) at day 7, the relative risk of hospitalization was 0.75 (95% CI, 0.32 to 1.77), and the difference in time to complete resolution of symptoms was -2 days (p = 0.38).
This study thus suggests that HCQ does not improve recovery from COVID-19, even in otherwise healthy adult patients with mild symptoms.
5.4.3 Prophylactic Administration of HCQ
An initial study of the possible prophylactic application of HCQ utilized a randomized, double-blind, placebo-controlled design to analyze the administration of HCQ prophylactically (1060).
Asymptomatic adults in the United States and Canada who had been exposed to SARS-CoV-2 within the past four days were enrolled in an online study to evaluate whether administration of HCQ over five days influenced the probability of developing COVID-19 symptoms over a 14-day period.
Of the participants, 414 received HCQ and 407 received a placebo.
No significant difference in the rate of symptomatic illness was observed between the two groups (11.8% HCQ, 14.3% placebo, p = 0.35).
The HCQ condition was associated with side effects, with 40.1% of patients reporting side effects compared to 16.8% in the control group (p < 0.001).
However, likely due to the high enrollment of healthcare workers (66% of participants) and the well-known side effects associated with HCQ, a large number of participants were able to correctly identify whether they were receiving HCQ or a placebo (46.5% and 35.7%, respectively).
Furthermore, due to a lack of availability of diagnostic testing, only 20 of the 107 cases were confirmed with a PCR-based test to be positive for SARS-CoV-2.
The rest were categorized as “probable” or “possible” cases by a panel of four physicians who were blind to the treatment status.
One possible confounder is that a patient presenting one or more symptoms, which included diarrhea, was defined as a “possible” case, but diarrhea is also a common side effect of HCQ.
Additionally, four of the twenty PCR-confirmed cases did not develop symptoms until after the observation period had completed, suggesting that the 14-day trial period may not have been long enough or that some participants also encountered secondary exposure events.
Finally, in addition to the young age of the participants in this study, which ranged from 32 to 51, there were possible impediments to generalization introduced by the selection process, as 2,237 patients who were eligible but had already developed symptoms by day 4 were enrolled in a separate study.
It is therefore likely that asymptomatic cases were over-represented in this sample, which would not have been detected based on the diagnostic criteria used.
Therefore, while this study does represent the first effort to conduct a randomized, double-blind, placebo-controlled investigation of HCQ’s effect on COVID-19 prevention after SARS-CoV-2 exposure in a large sample, the lack of PCR tests and several other design flaws significantly impede interpretation of the results.
However, in line with the results from therapeutic studies, once again no evidence was found suggesting an effect of HCQ against COVID-19.
A second study (1061) examined the effect of administering HCQ to healthcare workers as a pre-exposure prophylactic.
The primary outcome assessed was the conversion from SARS-CoV-2 negative to SARS-CoV-2 positive status over the 8 week study period.
This study was also randomized, double-blind, and placebo-controlled, and it sought to address some of the limitations of the first prophylactic study.
The goal was to enroll 200 healthcare workers, preferentially those working with COVID-19 patients, at two hospitals within the University of Pennsylvania hospital system in Philadelphia, PA.
Participants were randomized 1:1 to receive either 600 mg of HCQ daily or a placebo, and their SARS-CoV-2 infection status and antibody status were assessed using RT-PCR and serological testing, respectively, at baseline, 4 weeks, and 8 weeks following the beginning of the treatment period.
The statistical design of the study accounted for interim analyses at 50 and 100 participants in case efficacy or futility of HCQ for prophylaxis became clear earlier than completion of enrollment.
The 139 individuals enrolled comprised a study population that was fairly young (average age 33) and made of largely of people who were white, women, and without pre-existing conditions.
At the second interim analysis, more individuals in the treatment group than the control group had contracted COVID-19 (4 versus 3), causing the estimated z-score to fall below the pre-established threshold for futility.
As a result, the trial was terminated early, offering additional evidence against the use of HCQ for prophylaxis.
5.4.4 Summary of HCQ/CQ Research Findings
Early in vitro evidence indicated that HCQ could be an effective therapeutic against SARS-CoV-2 and COVID-19, leading to significant media attention and public interest in its potential as both a therapeutic and prophylactic.
Initially it was hypothesized that CQ/HCQ might be effective against SARS-CoV-2 in part because CQ and HCQ have both been found to inhibit the expression of CD154 in T-cells and to reduce TLR signaling that leads to the production of pro-inflammatory cytokines (1062).
Clinical trials for COVID-19 have more often used HCQ rather than CQ because it offers the advantages of being cheaper and having fewer side effects than CQ.
However, research has not found support for a positive effect of HCQ on COVID-19 patients.
Multiple clinical studies have already been carried out to assess HCQ as a therapeutic agent for COVID-19, and many more are in progress.
To date, none of these studies have used randomized, double-blind, placebo-controlled designs with a large sample size, which would be the gold standard.
Despite the design limitations (which would be more likely to produce false positives than false negatives), initial optimism about HCQ has largely dissipated.
The most methodologically rigorous analysis of HCQ as a prophylactic (1060) found no significant differences between the treatment and control groups, and the WHO’s global Solidarity trial similarly reported no effect of HCQ on mortality (811).
Thus, HCQ/CQ are not likely to be effective therapeutic or prophylactic agents against COVID-19.
One case study identified drug-induced phospholipidosis as the cause of death for a COVID-19 patient treated with HCQ (984), suggesting that in some cases, the proposed mechanism of action may ultimately be harmful.
Additionally, one study identified an increased risk of mortality in older men receiving HCQ, and administration of HCQ and HCQ + AZ did not decrease the use of mechanical ventilation in these patients (1058).
HCQ use for COVID-19 could also lead to shortages for anti-malarial or anti-rheumatic use, where it has documented efficacy.
Despite significant early attention, these drugs appear to be ineffective against COVID-19.
Several countries have now removed CQ/HCQ from their SOC for COVID-19 due to the lack of evidence of efficacy and the frequency of adverse effects.
5.5 ACE Inhibitors and Angiotensin II Receptor Blockers
Several clinical trials testing the effects of ACEIs or ARBs on COVID-19 outcomes are ongoing (1063–1069).
Clinical trials are needed because the findings of the various observational studies bearing on this topic cannot be interpreted as indicating a protective effect of the drug (1070, 1071).
Two analyses (1063, 1069) have reported no effect of continuing or discontinuing ARBs and ACEIs on patients admitted to the hospital for COVID-19.
The first, known as REPLACE COVID (874), was a randomized, open-label study that enrolled patients who were admitted to the hospital for COVID-19 and were taking an ACEI at the time of admission.
They enrolled 152 patients at 20 hospitals across seven countries and randomized them into two arms, continuation (n=75) and discontinuation (n=77).
The primary outcome evaluated was a global rank score that integrated several dimensions of illness.
The components of this global rank score, such as time to death and length of mechanical ventilation, were evaluated as secondary endpoints.
This analysis reported no differences between the two groups in the primary or any of the secondary outcomes.
Similarly, a second study (875) used a randomized, open-label design to examine the effects of continuing versus discontinuing ARBs and ACEIs on patients hospitalized for mild to moderate COVID-19 at 29 hospitals in Brazil.
This study enrolled 740 patients but had to exclude one trial site from all analyses due to the discovery of violations of Good Clinical Trial practice and data falsification.
After this exclusion, 659 patients remained, with 334 randomized to discontinuation and 325 to continuation.
In this study, the primary endpoint analyzed was the number of days that patients were alive and not hospitalized within 30 days of enrollment.
The secondary outcomes included death (including in-hospital death separately), number of days hospitalized, and specific clinical outcomes such as heart failure or stroke.
Once again, no significant differences were found between the two groups.
Initial studies of randomized interventions therefore suggest that ACEIs and ARBs are unlikely to affect COVID-19 outcomes.
These results are also consistent with findings from observational studies (summarized in (874)).
Additional information about ACE2, observational studies of ACEIs and ARBs in COVID-19, and clinical trials on this topic have been summarized (1072).
Therefore, despite the promising potential mechanism, initial results have not provided support for ACEIs and ARBs as therapies for COVID-19.
5.6 Tocilizumab
Human IL-6 is a 26-kDa glycoprotein that consists of 184 amino acids and contains two potential N-glycosylation sites and four cysteine residues.
It binds to a type I cytokine receptor (IL-6Rα or glycoprotein 80) that exists in both membrane-bound (IL-6Rα) and soluble (sIL-6Rα) forms (1073).
It is not the binding of IL-6 to the receptor that initiates pro- and/or anti-inflammatory signaling, but rather the binding of the complex to another subunit, known as IL-6Rβ or glycoprotein 130 (gp130) (1073, 1074).
Unlike membrane-bound IL-6Rα, which is only found on hepatocytes and some types of leukocytes, gp130 is found on most cells (1075).
When IL-6 binds to sIL-6Rα, the complex can then bind to a gp130 protein on any cell (1075).
The binding of IL-6 to IL-6Rα is termed classical signaling, while its binding to sIL-6Rα is termed trans-signaling (1075–1077).
These two signaling processes are thought to play different roles in health and illness.
For example, trans-signaling may play a role in the proliferation of mucosal T-helper TH2 cells associated with asthma, while an earlier step in this proliferation process may be regulated by classical signaling (1075).
Similarly, IL-6 is known to play a role in Crohn’s Disease via trans-, but not classical, signaling (1075).
Both classical and trans-signaling can occur through three independent pathways: the Janus-activated kinase-STAT3 pathway, the Ras/Mitogen-Activated Protein Kinases pathway and the Phosphoinositol-3 Kinase/Akt pathway (1073).
These signaling pathways are involved in a variety of different functions, including cell type differentiation, immunoglobulin synthesis, and cellular survival signaling pathways, respectively (1073).
The ultimate result of the IL-6 cascade is to direct transcriptional activity of various promoters of pro-inflammatory cytokines, such as IL-1, TFN, and even IL-6 itself, through the activity of NF-κB (1073).
IL-6 synthesis is tightly regulated both transcriptionally and post-transcriptionally, and it has been shown that viral proteins can enhance transcription of the IL-6 gene by strengthening the DNA-binding activity between several transcription factors and IL-6 gene-cis-regulatory elements (1078).
Therefore, drugs inhibiting the binding of IL-6 to IL-6Rα or sIL-6Rα are of interest for combating the hyperactive inflammatory response characteristic of cytokine release syndrome (CRS) and cytokine storm syndrome (CSS).
TCZ is a humanized monoclonal antibody that binds both to the insoluble and soluble receptor of IL-6, providing de facto inhibition of the IL-6 immune cascade.
Interest in TCZ as a possible treatment for COVID-19 was piqued by early evidence indicating that COVID-19 deaths may be induced by the hyperactive immune response, often referred to as CRS or CSS (83), as IL-6 plays a key role in this response (150).
The observation of elevated IL-6 in patients who died relative to those who recovered (83) could reflect an over-production of proinflammatory interleukins, suggesting that TCZ could potentially palliate some of the most severe symptoms of COVID-19 associated with increased cytokine production.
This early interest in TCZ as a possible treatment for COVID-19 was bolstered by a very small retrospective study in China that examined 20 patients with severe symptoms in early February 2020 and reported rapid improvement in symptoms following treatment with TCZ (893).
Subsequently, a number of retrospective studies have been conducted in several countries.
Many studies use a retrospective, observational design, where they compare outcomes for COVID-19 patients who received TCZ to those who did not over a set period of time.
For example, one of the largest retrospective, observational analyses released to date (888), consisting of 1,351 patients admitted to several care centers in Italy, compared the rates at which patients who received TCZ died or progressed to invasive medical ventilation over a 14-day period compared to patients receiving only SOC.
Under this definition, SOC could include other drugs such as HCQ, azithromycin, lopinavir-ritonavir or darunavir-cobicistat, or heparin.
While this study was not randomized, a subset of patients who were eligible to receive TCZ were unable to obtain it due to shortages; however, these groups were not directly compared in the analysis.
After adjusting for variables such as age, sex, and SOFA (sequential organ failure assessment) score, they found that patients treated with TCZ were less likely to progress to invasive medical ventilation and/or death (adjusted HR = 0.61, CI 0.40-0.92, p = 0.020); analysis of death and ventilation separately suggests that this effect may have been driven by differences in the death rate (20% of control versus 7% of TCZ-treated patients).
The study reported particular benefits for patients whose PaO2/FiO2 ratio, also known as the Horowitz Index for Lung Function, fell below a 150 mm Hg threshold.
They found no differences between groups administered subcutaneous versus intravenous TCZ.
Another retrospective observational analysis of interest examined the charts of patients at a hospital in Connecticut, USA where 64% of all 239 COVID-19 patients in the study period were administered TCZ based on assignment by a standardized algorithm (889).
They found that TCZ administration was associated with more similar rates of survivorship in patients with severe versus nonsevere COVID-19 at intake, defined based on the amount of supplemental oxygen needed.
They therefore proposed that their algorithm was able to identify patients presenting with or likely to develop CRS as good candidates for TCZ.
This study also reported higher survivorship in Black and Hispanic patients compared to white patients when adjusted for age.
The major limitation with interpretation for these studies is that there may be clinical characteristics that influenced medical practitioners decisions to administer TCZ to some patients and not others.
One interesting example therefore comes from an analysis of patients at a single hospital in Brescia, Italy, where TCZ was not available for a period of time (890).
This study compared COVID-19 patients admitted to the hospital before and after March 13, 2020, when the hospital received TCZ.
Therefore, patients who would have been eligible for TCZ prior to this arbitrary date did not receive it as treatment, making this retrospective analysis something of a natural experiment.
Despite this design, demographic factors did not appear to be consistent between the two groups, and the average age of the control group was older than the TCZ group.
The control group also had a higher percentage of males and a higher incidence of comorbidities such as diabetes and heart disease.
All the same, the multivariate HR, which adjusted for these clinical and demographic factors, found a significant difference between survival in the two groups (HR=0.035, CI=0.004-0.347, p = 0.004).
The study reported improvement of survival outcomes after the addition of TCZ to the SOC regime, with 11 of 23 patients (47.8%) admitted prior to March 13th dying compared to 2 of 62 (3.2%) admitted afterwards (HR=0.035; 95% CI, 0.004 to 0.347; p = 0.004).
They also reported a reduced progression to mechanical ventilation in the TCZ group.
However, this study also holds a significant limitation: the time delay between the two groups means that knowledge about how to treat the disease likely improved over this timeframe as well.
All the same, the results of these observational retrospective studies provide support for TCZ as a pharmaceutical of interest for follow-up in clinical trials.
Other retrospective analyses have utilized a case-control design to match pairs of patients with similar baseline characteristics, only one of whom received TCZ for COVID-19.
In one such study, TCZ was significantly associated with a reduced risk of progression to intensive care unit (ICU) admission or death (891).
This study examined only 20 patients treated with TCZ (all but one of the patients treated with TCZ in the hospital during the study period) and compared them to 25 patients receiving SOC.
For the combined primary endpoint of death and/or ICU admission, only 25% of patients receiving TCZ progressed to an endpoint compared to 72% in the SOC group (p = 0.002, presumably based on a chi-square test based on the information provided in the text).
When the two endpoints were examined separately, progression to invasive medical ventilation remained significant (32% SOC compared to 0% TCZ, p = 0.006) but not for mortality (48% SOC compared to 25% TCZ, p = 0.066).
In contrast, a study that compared 96 patients treated with TCZ to 97 patients treated with SOC only in New York City found that differences in mortality did not differ between the two groups, but that this difference did become significant when intubated patients were excluded from the analysis (892).
Taken together, these findings suggest that future clinical trials of TCZ may want to include intubation as an endpoint.
However, these studies should be approached with caution, not only because of the small number of patients enrolled and the retrospective design, but also because they performed a large number of statistical tests and did not account for multiple hypothesis testing.
In general, caution must be exercised when interpreting subgroup analyses after a primary combined endpoint analysis.
These last findings highlight the need to search for a balance between impairing a harmful immune response, such as the one generated during CRS/CSS, and preventing the worsening of the clinical picture of the patients by potential new viral infections.
Early meta-analyses and systematic reviews have investigated the available data about TCZ for COVID-19.
One meta-analysis (1079) evaluated 19 studies published or released as preprints prior to July 1, 2020 and found that the overall trends were supportive of the frequent conclusion that TCZ does improve survivorship, with a significant HR of 0.41 (p < 0.001).
This trend improved when they excluded studies that administered a steroid alongside TCZ, with a significant HR of 0.04 (p < 0.001).
They also found some evidence for reduced invasive ventilation or ICU admission, but only when excluding all studies except a small number whose estimates were adjusted for the possible bias introduced by the challenges of stringency during the enrollment process.
A systematic analysis of sixteen case-control studies of TCZ estimated an odds ratio of mortality of 0.453 (95% CI 0.376–0.547, p < 0.001), suggesting possible benefits associated with TCZ treatment (1080).
Although these estimates are similar, it is important to note that they are drawing from the same literature and are therefore likely to be affected by the same potential biases in publication.
A different systematic review of studies investigating TCZ treatment for COVID-19 analyzed 31 studies that had been published or released as pre-prints and reported that none carried a low risk of bias (1081).
Therefore, the present evidence is not likely to be sufficient for conclusions about the efficacy of TCZ.
On February 11, 2021, a preprint describing the first randomized control trial of TCZ was released as part of the RECOVERY trial (894).
Of the 21,550 patients enrolled in the RECOVERY trial at the time, 4,116 adults hospitalized with COVID-19 across the 131 sites in the United Kingdom were assigned to the arm of the trial evaluating the effect of TCZ.
Among them, 2,022 were randomized to receive TCZ and 2,094 were randomized to SOC, with 79% of patients in each group available for analysis at the time that the initial report was released.
The primary outcome measured was 28-day mortality, and TCZ was found to reduce 28-day mortality from 33% of patients receiving SOC alone to 29% of those receiving TCZ, corresponding to a rate ratio of 0.86 (95% CI 0.77-0.96; p = 0.007).
TCZ was also significantly associated with the probability of hospital discharge within 28 days for living patients, which was 47% in the SOC group and 54% in the TCZ group (rate ratio 1.22, 95% CI 1.12-1.34, p < 0.0001).
A potential statistical interaction between TCZ and corticosteroids was observed, with the combination providing greater mortality benefits than TCZ alone, but the authors note that caution is advisable in light of the number of statistical tests conducted.
Combining the RECOVERY trial data with data from seven smaller randomized control trials indicates that TCZ is associated with a 13% reduction in 28-day mortality (rate ratio 0.87, 95% CI 0.79-0.96, p = 0.005) (894).
There are possible risks associated with the administration of TCZ for COVID-19.
TCZ has been used for over a decade to treat RA (1082), and a recent study found the drug to be safe for pregnant and breastfeeding women (1083).
However, TCZ may increase the risk of developing infections (1082), and RA patients with chronic hepatitis B infections had a high risk of hepatitis B virus reactivation when TCZ was administered in combination with other RA drugs (1084).
As a result, TCZ is contraindicated in patients with active infections such as tuberculosis (1085).
Previous studies have investigated, with varying results, a possible increased risk of infection in RA patients administered TCZ (1086, 1087), although another study reported that the incidence rate of infections was higher in clinical practice RA patients treated with TCZ than in the rates reported by clinical trials (1088).
In the investigation of 544 Italian COVID-19 patients, the group treated with TCZ was found to be more likely to develop secondary infections, with 24% compared to 4% in the control group (p < 0.0001) (888).
Reactivation of hepatitis B and herpes simplex virus 1 was also reported in a small number of patients in this study, all of whom were receiving TCZ.
A July 2020 case report described negative outcomes of two COVID-19 patients after receiving TCZ, including one death; however, both patients were intubated and had entered septic shock prior to receiving TCZ (1089), likely indicating a severe level of cytokine production.
Additionally, D-dimer and sIL2R levels were reported by one study to increase in patients treated with TCZ, which raised concerns because of the potential association between elevated D-dimer levels and thrombosis and between sIL2R and diseases where T-cell regulation is compromised (889).
An increased risk of bacterial infection was also identified in a systematic review of the literature, based on the unadjusted estimates reported (1079).
In the RECOVERY trial, however, only three out of 2,022 participants in the group receiving TCZ developed adverse reactions determined to be associated with the intervention, and no excess deaths were reported (894).
TCZ administration to COVID-19 patients is not without risks and may introduce additional risk of developing secondary infections; however, while caution may be prudent when treating patients who have latent viral infections, the results of the RECOVERY trial indicate that adverse reactions to TCZ are very rare among COVID-19 patients broadly.
In summary, approximately 33% of hospitalized COVID-19 patients develop ARDS (1090), which is caused by an excessive early response of the immune system which can be a component of CRS/CSS (889, 1085).
This overwhelming inflammation is triggered by IL-6.
TCZ is an inhibitor of IL-6 and therefore may neutralize the inflammatory pathway that leads to the cytokine storm.
The mechanism suggests TCZ could be beneficial for the treatment of COVID-19 patients experiencing excessive immune activity, and the RECOVERY trial reported a reduction in 28-day mortality.
Interest in TCZ as a treatment for COVID-19 was also supported by two meta-analyses (1079, 1091), but a third meta-analysis found that all of the available literature at that time carried a risk of bias (1081).
Additionally, different studies used different dosages, number of doses, and methods of administration.
Ongoing research may be needed to optimize administration of TCZ (1092), although similar results were reported by one study for intravenous and subcutaneous administration (888).
Clinical trials that are in progress are likely to provide additional insight into the effectiveness of this drug for the treatment of COVID-19 along with how it should be administered.
5.7 Interferons
IFNs are a family of cytokines critical to activating the innate immune response against viral infections.
Interferons are classified into three categories based on their receptor specificity: types I, II and III (150).
Specifically, IFNs I (IFN-𝛼 and 𝛽) and II (IFN-𝛾) induce the expression of antiviral proteins (1093).
Among these IFNs, IFN-𝛽 has already been found to strongly inhibit the replication of other coronaviruses, such as SARS-CoV-1, in cell culture, while IFN-𝛼 and 𝛾 were shown to be less effective in this context (1093).
There is evidence that patients with higher susceptibility to ARDS indeed show deficiency in IFN-𝛽.
For instance, infection with other coronaviruses impairs IFN-𝛽 expression and synthesis, allowing the virus to escape the innate immune response (1094).
On March 18 2020, Synairgen plc received approval to start a phase II trial for SNG001, an IFN-𝛽-1a formulation to be delivered to the lungs via inhalation (901).
SNG001, which contains recombinant interferon beta-1a, was previously shown to be effective in reducing viral load in an in vivo model of swine flu and in vitro models of other coronavirus infections (1095).
In July 2020, a press release from Synairgen stated that SNG001 reduced progression to ventilation in a double-blind, placebo-controlled, multi-center study of 101 patients with an average age in the late 50s (902).
These results were subsequently published in November 2020 (903).
The study reports that the participants were assigned at a ratio of 1:1 to receive either SNG001 or a placebo that lacked the active compound, by inhalation for up to 14 days.
The primary outcome they assessed was the change in patients’ score on the WHO Ordinal Scale for Clinical Improvement (OSCI) at trial day 15 or 16.
SNG001 was associated with an odds ratio of improvement on the OSCI scale of 2.32 (95% CI 1.07 – 5.04, p = 0.033) in the intention-to-treat analysis and 2.80 (95% CI 1.21 – 6.52, p = 0.017) in the per-protocol analysis, corresponding to significant improvement in the SNG001 group on the OSCI at day 15/16.
Some of the secondary endpoints analyzed also showed differences: at day 28, the OR for clinical improvement on the OSCI was 3.15 (95% CI 1.39 – 7.14, p = 0.006), and the odds of recovery at day 15/16 and at day 28 were also significant between the two groups.
Thus, this study suggested that IFN-𝛽1 administered via SNG001 may improve clinical outcomes.
In contrast, the WHO Solidarity trial reported no significant effect of IFN-β-1a on patient survival during hospitalization (811).
Here, the primary outcome analyzed was in-hospital mortality, and the rate ratio for the two groups was 1.16 (95% CI, 0.96 to 1.39; p = 0.11) administering IFN-β-1a to 2050 patients and comparing their response to 2,050 controls.
However, there are a few reasons that the different findings of the two trials might not speak to the underlying efficacy of this treatment strategy.
One important consideration is the stage of COVID-19 infection analyzed in each study.
The Synairgen trial enrolled only patients who were not receiving invasive ventilation, corresponding to a less severe stage of disease than many patients enrolled in the SOLIDARITY trial, as well as a lower overall rate of mortality (1096).
Additionally, the methods of administration differed between the two trials, with the SOLIDARITY trial administering IFN-β-1a subcutaneously (1096).
The differences in findings between the studies suggests that the method of administration might be relevant to outcomes, with nebulized IFN-β-1a more directly targeting receptors in the lungs.
A trial that analyzed the effect of subcutaneously administered IFN-β-1a on patients with ARDS between 2015 and 2017 had also reported no effect on 28-day mortality (1097), while a smaller study analyzing the effect of subcutaneous IFN administration did find a significant improvement in 28-day mortality for COVID-19 (1098).
At present, several ongoing clinical trials are investigating the potential effects of IFN-β-1a, including in combination with therapeutics such as remdesivir (1099) and administered via inhalation (901).
Thus, as additional information becomes available, a more detailed understanding of whether and under which circumstances IFN-β-1a is beneficial to COVID-19 patients should develop.
5.8 Potential Avenues of Interest for Therapeutic Development
Given what is currently known about these therapeutics for COVID-19, a number of related therapies beyond those explored above may also prove to be of interest.
For example, the demonstrated benefit of dexamethasone and the ongoing potential of tocilizumab for treatment of COVID-19 suggests that other anti-inflammatory agents might also hold value for the treatment of COVID-19.
Current evidence supporting the treatment of severe COVID-19 with dexamethasone suggests that the need to curtail the cytokine storm inflammatory response transcends the risks of immunosuppression, and other anti-inflammatory agents may therefore benefit patients in this phase of the disease.
While dexamethasone is considered widely available and generally affordable, the high costs of biologics such as tocilizumab therapy may present obstacles to wide-scale distribution of this drug if it proves of value.
At the doses used for RA patients, the cost for tocilizumab ranges from $179.20 to $896 per dose for the IV form and $355 for the pre-filled syringe (1100).
Several other anti-inflammatory agents used for the treatment of autoimmune diseases may also be able to counter the effects of the cytokine storm induced by the virus, and some of these, such as cyclosporine, are likely to be more cost-effective and readily available than biologics (1101).
While tocilizumab targets IL-6, several other inflammatory markers could be potential targets, including TNF-α.
Inhibition of TNF-α by a compound such as Etanercept was previously suggested for treatment of SARS-CoV-1 (1102) and may be relevant for SARS-CoV-2 as well.
Another anti-IL-6 antibody, sarilumab, is also being investigated (1103, 1104).
Baricitinib and other small molecule inhibitors of the Janus-activated kinase pathway also curtail the inflammatory response and have been suggested as potential options for SARS-CoV-2 infections (1105).
Baricitinib, in particular, may be able to reduce the ability of SARS-CoV-2 to infect lung cells (1106).
Clinical trials studying baricitinib in COVID-19 have already begun in the US and in Italy (1107, 1108).
Identification and targeting of further inflammatory markers that are relevant in SARS-CoV-2 infection may be of value for curtailing the inflammatory response and lung damage.
In addition to immunosuppressive treatments, which are most beneficial late in disease progression, much research is focused on identifying therapeutics for early-stage patients.
For example, although studies of HCQ have not supported the early theory-driven interest in this antiviral treatment, alternative compounds with related mechanisms may still have potential.
Hydroxyferroquine derivatives of HCQ have been described as a class of bioorganometallic compounds that exert antiviral effects with some selectivity for SARS-CoV-1 in vitro(1109).
Future work could explore whether such compounds exert antiviral effects against SARS-CoV-2 and whether they would be safer for use in COVID-19.
Another potential approach is the development of antivirals, which could be broad-spectrum, specific to coronaviruses, or targeted to SARS-CoV-2.
Development of new antivirals is complicated by the fact that none have yet been approved for human coronaviruses.
Intriguing new options are emerging, however.
Beta-D-N4-hydroxycytidine is an orally bioavailable ribonucleotide analog showing broad-spectrum activity against RNA viruses, which may inhibit SARS-CoV-2 replication in vitro and in vivo in mouse models of HCoVs (1110).
A range of other antivirals are also in development.
Development of antivirals will be further facilitated as research reveals more information about the interaction of SARS-CoV-2 with the host cell and host cell genome, mechanisms of viral replication, mechanisms of viral assembly, and mechanisms of viral release to other cells; this can allow researchers to target specific stages and structures of the viral life cycle.
Finally, antibodies against viruses, also known as antiviral monoclonal antibodies, could be an alternative as well and are described in detail in an above section.
The goal of antiviral antibodies is to neutralize viruses through either cell-killing activity or blocking of viral replication (1111).
They may also engage the host immune response, encouraging the immune system to hone in on the virus.
Given the cytokine storm that results from immune system activation in response to the virus, which has been implicated in worsening of the disease, a neutralizing antibody (nAb) may be preferable.
Upcoming work may explore the specificity of nAbs for their target, mechanisms by which the nAbs impede the virus, and improvements to antibody structure that may enhance the ability of the antibody to block viral activity.
Some research is also investigating potential therapeutics and prophylactics that would interact with components of the innate immune response.
For example, TLRs are pattern recognition receptors that recognize pathogen- and damage-associated molecular patterns and contribute to innate immune recognition and, more generally, promotion of both the innate and adaptive immune responses (147).
In mouse models, poly(I:C) and CpG, which are agonists of Toll-like receptors TLR3 and TLR9, respectively, showed protective effects when administered prior to SARS-CoV-1 infection (1112).
Therefore, TLR agonists hold some potential for broad-spectrum prophylaxis.
6 Application of Traditional Vaccine Development Strategies to SARS-CoV-2
6.1 Abstract
Over the past 150 years, vaccines have revolutionized the relationship between people and disease.
During the COVID-19 pandemic, technologies such as mRNA vaccines have received attention due to their novelty and successes.
However, more traditional vaccine development platforms have also yielded important tools in the worldwide fight against the SARS-CoV-2 virus.
A variety of approaches have been used to develop COVID-19 vaccines that are now authorized for use in countries around the world.
In this review, we highlight strategies that focus on the viral capsid and outwards, rather than on the nucleic acids inside.
These approaches fall into two broad categories: whole-virus vaccines and subunit vaccines.
Whole-virus vaccines use the virus itself, either in an inactivated or attenuated state.
Subunit vaccines contain instead an isolated, immunogenic component of the virus.
Here, we highlight vaccine candidates that apply these approaches against SARS-CoV-2 in different ways.
In a companion manuscript, we review the more recent and novel development of nucleic-acid based vaccine technologies.
We further consider the role that these COVID-19 vaccine development programs have played in prophylaxis at the global scale.
Well-established vaccine technologies have proved especially important to making vaccines accessible in low- and middle-income countries.
Vaccine development programs that use established platforms have been undertaken in a much wider range of countries than those using nucleic-acid-based technologies, which have been led by wealthy Western countries.
Therefore, these vaccine platforms, though less novel from a biotechnological standpoint, have proven to be extremely important to the management of SARS-CoV-2.
6.2 Importance
As of March 9, 2023, there have been over 676,570,149 SARS-CoV-2 cases, and the virus has taken the lives of at least 6,881,802 people globally (732).
The development, production, and distribution of vaccines is imperative to saving lives, preventing illness, and reducing the economic and social burdens caused by the COVID-19 pandemic.
Vaccines that use cutting-edge biotechnology have played an important role in mitigating the effects of SARS-CoV-2.
However, more traditional methods of vaccine development that were refined throughout the twentieth century have been especially critical to increasing vaccine access worldwide.
Effective deployment is necessary to reducing the susceptibility of the world’s population, which is especially important in light of emerging variants.
In this review, we discuss the safety, immunogenicity, and distribution of vaccines developed using established technologies.
In a separate review, we describe the vaccines developed using nucleic acid-based vaccine platforms.
From the current literature, it is clear that the well-established vaccine technologies are also highly effective against SARS-CoV-2 and are being used to address the challenges of COVID-19 globally, including in low- and middle-income countries.
This worldwide approach is critical for reducing the devastating impact of SARS-CoV-2.
6.3 Introduction
The development of vaccines is widely considered one of the most important medical advances in human history.
Over the past 150 years, several approaches to vaccination have been developed and refined (1113).
The COVID-19 pandemic has produced unusual circumstances compared to past health crises, leading to differences in vaccine development strategies.
One way in which the COVID-19 pandemic differs from prior global health crises is that the SARS-CoV-2 viral genome was sequenced, assembled, and released very early in the course of the pandemic (January 2020).
This genomic information has informed the biomedical response to this novel pathogen across several dimensions (1, 3).
All the same, vaccines have been developed since long before the concept of a virus or a viral genome was known, and as early as September 2020, there were over 180 vaccine candidates against SARS-CoV-2 in development, many of which employed more traditional vaccine technologies (1114).
However, public attention in the United States and elsewhere has largely focused on vaccine development platforms that use new technologies, especially mRNA vaccines.
We review vaccine technologies used for SARS-CoV-2 in two parts: here, the application of established vaccine development platforms to SARS-CoV-2, and separately, novel nucleic acid-based approaches (6).
Understanding vaccine development programs that are using well-established technologies is important for a global perspective on COVID-19.
As of May 3, 2023, 50 SARS-CoV-2 vaccines have been approved for use in at least one country (1115).
A resource tracking the distribution of 28 vaccines indicates that, as of May 31, 2023, 13.0 billion doses have been administered across 223 countries (1116).
Many of these vaccines use platforms that do not require information about the viral genome, with 20 developed using subunit and 11 using whole-virus approaches (1115).
The types of vaccines available varies widely throughout the world, as the process of developing and deploying a vaccine is complex and often requires coordination between government, industry, academia, and philanthropic entities (1117).
Another difference between prior global health crises and the COVID-19 pandemic is the way that vaccines are evaluated.
A vaccine’s success is often discussed in terms of vaccine efficacy (VE), which describes the protection conferred during clinical trials (1118).
The real-world protection offered by a vaccine is referred to as its effectiveness (1118).
Additionally, protection can mean different things in different contexts.
In general, the goal of a vaccine is to prevent disease, especially severe disease, rather than infection itself.
As a proxy for VE, vaccine developers often test their candidates for serum neutralizing activity, which has been proposed as a biomarker for adaptive immunity in other respiratory illnesses (1119).
The duration and intensity of the COVID-19 pandemic has made it possible to test multiple vaccines in phase III trials, where the effect of the vaccines on a cohort’s likelihood of contracting SARS-CoV-2 can be evaluated, whereas this has not always been feasible for other infectious diseases.
In some cases (e.g., SARS), the pathogen has been controlled before vaccine candidates were available, while in others (e.g., MERS), the scale of the epidemic has been smaller.
Vaccine development is traditionally a slow process, but the urgency of the COVID-19 pandemic created an atypical vaccine development ecosystem where fast development and production was prioritized.
Estimates of VE have been released for many vaccine candidates across a number of technology types based on phase III trial data.
However, efficacy is not a static value, and both trial efficacy and real-world effectiveness can vary across location and over time.
Shifts in effectiveness in particular have been an especially heightened topic of concern since late 2021 given the potential for variants of SARS-CoV-2 to influence VE.
Due to viral evolution, vaccine developers are in an arms race with a pathogen that benefits from mutations that reduce its susceptibility to adaptive immunity.
The evolution of several variants of concern (VOC) presents significant challenges for vaccines developed based on the index strain identified in Wuhan in late 2019.
We discuss these variants in depth elsewhere (341).
To date, the most significant VOC identified are Alpha (2020), Beta (2020), Gamma (2020), Delta (2021), and Omicron (2021), with various subvariants of Omicron being the most recently identified (2022).
The relative timing of studies relative to dominant VOC in the region where participants are recruited is important context for a complete picture of efficacy.
Therefore, the efficacy and/or effectiveness of vaccines in the context of these variants is discussed where information is available.
Beyond the variability introduced by time and geography, efficacy within a trial and effectiveness in the real-world setting can also differ due to cohort differences.
Patients participating in a clinical trial are likely to receive more medical oversight, resulting in better follow-up, adherence, and patient engagement (1120).
Additionally, the criteria for participant inclusion in a trial often bias trials towards selection of younger, healthier individuals (1121).
The ability of an RCT to accurately assess safety can be biased by the fact that a clinical trial might not reveal rare adverse events (AEs) that might become apparent on a larger scale (1121).
Therefore, while clinical trials are the gold standard for evaluating vaccines for COVID-19, the results of these trials must be considered in a broader context when real-world data is available.
While the relationship between a vaccine and a pathogen is not static, the data clearly demonstrates that a variety of efficacious vaccines have been developed against SARS-CoV-2.
Here we discuss a selection of programs that use well-established vaccine biotechnologies.
These programs have been undertaken worldwide, in complement to the more cutting-edge approaches developed and distributed in the United States (U.S.), the European Union (E.U.), the United Kingdom (U.K.), and Russia (6).
In this review, we discuss vaccine development using two well-established technologies, whole-virus vaccination and subunit vaccination, and the role these technologies have played in the global response to the COVID-19 pandemic.
6.4 Whole-Virus Vaccines
Whole-virus vaccines have the longest history among vaccine development approaches.
Variolation, which is widely considered the first vaccination strategy in human history, is one example (1122, 1123).
Famously, variolation was employed against smallpox when healthy individuals were exposed to pus from an individual infected with what was believed to be either cowpox or horsepox (1122–1125).
This approach worked by inducing a mild case of a disease.
Therefore, while whole-virus vaccines confer adaptive immunity, they also raise safety concerns (1124, 1126, 1127).
As of 2005, most vaccines still used whole-virus platforms (1128), and these technologies remain valuable tools in vaccine development today (1113).
Whole virus vaccine candidates have been developed for COVID-19 using both live attenuated viruses and inactivated whole viruses.
6.4.1 Live-Attenuated Virus Vaccines
Live-attenuated virus vaccines (LAV), also known as replication-competent vaccines, use a weakened, living version of a disease-causing virus or a version of a virus that is modified to induce an immune response (1114).
Whether variolation is the first example of a LAV being used to induce immunity is debated (1113, 1126).
The first deliberate (albeit pathogen-naïve) attempt to develop an attenuated viral vaccine dates back to Louis Pasteur’s efforts in 1885 to inoculate a child against rabies (1129).
The next intentional LAVs were developed against the yellow fever virus in 1935 and influenza in 1936 (1130).
Early efforts in LAV development relied on either the identification of a related virus that was less virulent in humans (e.g., cowpox/horsepox or rotavirus vaccines) or the culturing of a virus in vitro(1113, 1124).
Today, a virus can be attenuated by passaging it in a foreign host until, due to selection pressure, the virus loses its efficacy in the original host.
Alternatively, selective gene deletion or codon deoptimization can be utilized to attenuate the virus (1114), or foreign antigens can be integrated into an attenuated viral vector (1131).
LAVs tend to be restricted to viral replication in the tissues around the location of inoculation (1130), and some can be administered intranasally (1114).
Today, LAVs are used globally to prevent diseases caused by viruses such as measles, mumps, rubella, polio, influenza, varicella zoster, and the yellow fever virus (1132).
There were attempts to develop LAVs against both SARS-CoV-1 and MERS-CoV (1133), but no vaccines were approved.
It is generally recognized that LAVs induce an immune response similar to natural infection, and they are favored because they induce long-lasting and robust immunity that can protect from disease.
This strong protective effect is induced in part by the immune response to the range of viral antigens available from LAV, which tend to be more immunogenic than those from non-replicating vaccines (1126, 1133, 1134).
6.4.2 LAV Vaccines and COVID-19
To date, LAVs have not been widely deployed against SARS-CoV-2 and COVID-19.
All the same, there are several COVID-19 LAV candidates in the early (preclinical/phase I) stages of investigation.
These candidates utilize different approaches.
Interestingly, several candidates (Meissa Vaccines’ MV-014-212 and Codagenix’s COVI-VAC, specifically) are administered intranasally, potentially improving accessibility by eliminating the need for sterile needles and reducing manufacturing costs, targeting conferring mucosal immunity, and reducing some issues related to vaccine hesitancy (1135, 1136).
Additionally, although no phase III trial data is available for LAV vaccine candidates, some manufacturers have proactively sought to respond to the emergence of VOC.
Therefore, the original approach to vaccination may still prove extremely advantageous in the high-tech landscape of COVID-19 vaccine development.
6.4.2.1 YF-S0
One candidate in the preclinical stage is YF-S0, a single-dose LAV developed at Belgium’s Katholieke Universiteit Leuven that uses live-attenuated yellow fever 17D (YF17D) as a vector for a noncleavable prefusion conformation of the spike antigen of SARS-CoV-2 (1131).
YF-S0 induced a robust immune response in three animal models and prevented SARS-CoV-2 infection in macaques and hamsters (1131).
Additionally, the protective effect of YF-S0 against VOC has been investigated in hamsters (1137).
Even for a small number of hamsters that developed breakthrough infections after exposure to the index strain or the Alpha variant, viral loads were very low (1137).
However, much higher rates of breakthrough infection and higher viral loads were observed when the hamsters were exposed to the Beta variant (1137).
Reduced seroconversion and nAb titers were also observed against the Beta and Gamma variants (1137).
As a result, a modified version of YF-S0, called YF-S0*, was developed to include a modified spike protein intended to increase immunogenicity by including the full spectrum of amino acids found in the Gamma VOC as well as stabilizing the S protein’s conformation (1137).
The updated vaccine was again tested in Syrian golden hamsters (1137).
No breakthrough infections were observed following vaccination with YF-S0* and exposure to the index strain and the Alpha, Beta, Gamma, and Delta variants (1137).
YF-S0* also reduced the infectious viral load in the lungs of several VOCs (Alpha, Beta, Gamma, and Delta) relative to a sham comparison (1137), and the likelihood of the Delta variant spreading to unvaccinated co-housed hamsters was significantly reduced by YF-S0* (1137).
The updated vaccine was also associated with the increased production of nAbs against the Omicron variant compared to YF-S0 (1137).
6.4.2.2 COVI-VAC
Other programs are developing codon deoptimized LAV candidates (1138–1140).
This approach follows the synthetic attenuated virus engineering (SAVE) strategy to select codon substitutions that are suboptimal for the virus (1140, 1141).
New York-based Codagenix and the Serum Institute of India reported a successful preclinical investigation (1140) of an intranasally administered deoptimized SARS-CoV-2 LAV known as COVI-VAC, and COVI-VAC entered phase I human trials and dosed its first participants in January 2021 (1139, 1142).
This vaccine is optimized through the removal of the furin cleavage site (see (1) for a discussion of this site’s importance) and deoptimization of 283 codons (1143).
The results of the COVI-VAC phase I human trials are expected soon (1142).
Other results suggest both potential benefits and risks to the COVI-VAC vaccine candidate.
Preclinical results suggest that the vaccine candidate may confer some protection against VOC even though it was designed based on the index strain: a poster reported that Syrian golden hamsters who received COVI-VAC were significantly less likely to lose weight following viral challenge with the Beta VOC (1143).
On the other hand, some concerns have arisen about the possibility of spillover from LAV vaccines.
A December 2022 study analyzed SARS-CoV-2 samples isolated from COVID-19 patients in India and identified two extremely similar sequences collected on June 30, 2020 that showed a high level of recombination relative to the dominant strains at the time (1144).
Comparing these samples to a database of SARS-CoV-2 sequences revealed they were most similar to the sequence used for COVI-VAC (1144).
Based on phylogenetic reconstruction, the authors argued that these SARS-CoV-2 isolates were most likely to have spilled over from COVI-VAC trials (1144).
If this was a case of spillover, the effect seems to have been limited, as these sequences were just two among over 1,600 analyzed.
However, these concerns may be one consideration in the development of LAV vaccines for COVID-19.
6.4.2.3 Meissa Vaccines MV-014-212
Another company, Meissa Vaccines in Kansas, U.S., which also develops vaccines for respiratory syncytial virus (RSV), has developed an intranasal live-attenuated chimeric vaccine MV-014-212 (1145).
Chimeric vaccines integrate genomic content from multiple viruses to create a more stable LAV (1146).
To develop a SARS-CoV-2 vaccine candidate, Meissa Vaccines built on their prior work developing RSV vaccines (1145).
A live attenuated recombinant strain of RSV previously investigated as a vaccine candidate was modified to replace two surface glycoproteins with a chimeric protein containing components of the SARS-CoV-2 Spike protein as well as the RSV fusion (F) protein (1145).
Preclinical results describing the intranasal administration of MV-014-212 to African green monkeys and mice indicated that the vaccine candidate produced neutralizing antibodies (nAb) as well as a cellular immune response to SARS-CoV-2 challenge, including the Alpha, Beta, and Delta VOC (1145).
Enrollment for phase I human trials began in March 2021 and recruitment is ongoing (1139, 1147).
6.4.2.4 Bacillus Calmette-Guerin Vaccines
Finally, Bacillus Calmette-Guerin (BCG) vaccines that use LAVs are being investigated for the prophylaxis of COVID-19 (see online Appendix (1148)).
The purpose of the BCG vaccine is to prevent tuberculosis, but non-specific effects against other respiratory illnesses have suggested a possible benefit against COVID-19 (1149).
However, a multicenter trial that randomly assigned participants 60 years and older to vaccination with BCG (n = 1,008) or placebo (n = 1,006) found that BCG vaccination had no effect on the incidence of SARS-CoV-2 or other respiratory infections over the course of 12 months (1150).
Despite these findings, BCG vaccination was associated with a stronger cytokine (specifically, IL-6) response following ex vivo stimulation of peripheral blood mononuclear cells in patients with no known history of COVID-19 (1150).
Additionally, SARS-CoV-2-positive individuals who had received the BCG vaccine one year prior showed increased immunoglobulin (Ig) responses to the SARS-CoV-2 spike protein and receptor binding domain (RBD) relative to individuals who had received a placebo vaccine (1150).
Currently, investigations of BCG vaccines against COVID-19 are being sponsored by institutes in Australia in collaboration with the Bill and Melinda Gates Foundation (1151) and by Texas A&M University in collaboration with numerous other U.S. institutions (1152).
6.4.2.5 Summary of LAV Vaccine Development
LAV vaccines for COVID-19 have not advanced as far in development as vaccines developed using other technologies.
As of December 2022, COVI-VAC was the only LAV vaccine candidate in phase III clinical trials (1153).
As a result, safety data is not yet available for human studies of these vaccines.
In general, though, safety concerns previously associated with LAV have been largely mitigated in the modern manufacturing process.
Manufacturers use safe and reliable methods to produce large quantities of vaccines once they have undergone rigorous preclinical studies and clinical trials to evaluate their safety and efficacy.
However, one remaining safety concern may be contributing to the relatively slow emergence of LAV candidates against COVID-19: they still may present risk to individuals who are immunocompromised (1154), which is an even greater concern when dealing with a novel virus and disease.
Additional data are needed to ascertain how this technology performs in the case of SARS-CoV-2 and whether rare cases of spillover have indeed occurred.
Additionally, modifications to the design of individual vaccine candidates may make this protection more robust as SARS-CoV-2 evolves, as the limited data about LAV performance against VOC suggests.
Despite the long and trusted history of LAV development, this vaccine strategy has not been favored against COVID-19, as other technologies have shown greater expediency and safety compared to the time-consuming nature of developing LAVs for a novel virus.
6.5 Inactivated Whole-Virus Vaccines
Inactivated whole-virus (IWV) vaccines are another well-established vaccine platform.
This platform uses full virus particles generally produced via cell culture that have been rendered non-infectious by chemical (i.e., formaldehyde or β-propiolactone (1155)) or physical (i.e., heat or ultraviolet radiation) means.
In general, these vaccines mimic the key properties of the virus that stimulate a robust immune response, but the risk of adverse reactions is reduced because the virus is inactivated and thus unable to replicate.
Though these viral particles are inactivated, they retain the capacity to prime the immune system.
The size of the viral particle makes it ideal for uptake by antigen-presenting cells, which leads to the stimulation of helper T-cells (1156).
Additionally, the array of epitopes on the surface of the virus increases antibody binding efficiency (1156).
The native conformation of the surface proteins, which is also important for eliciting an immune response, is preserved using these techniques (1157).
Membrane proteins, which support B-cell responses to surface proteins, are also induced by this method (1158).
IWV vaccines have been a valuable tool in efforts to control many viruses.
Some targets of IWV vaccines have included influenza viruses, poliovirus, and hepatitis A virus.
Inactivated vaccines can generally be generated relatively quickly once the pathogenic virus has been isolated and can be passaged in cell culture (1133, 1159).
During COVID-19, though the World Health Organization (WHO) has been slower to approve IWV vaccine candidates than those developed with nucleic acid-based technologies, IWV vaccine development was also fast.
In China, the first emergency use authorization (EUA) was granted to an IWV vaccine in July 2020, with full approval following that December (1160, 1161).
The fact that these vaccines have not received as much public attention (at least in Western media) as nucleic acid vaccines for SARS-CoV-2 may be due at least in part to the novelty of nucleic acid vaccine technologies (1162), which are more modular and immunogenic (6).
Past applications to human coronaviruses (HCoV) have focused predominantly on SARS-CoV-1.
Preclinical studies have demonstrated that IWV SARS-CoV-1 vaccine candidates elicited immune responses in vivo.
These vaccines generated nAb titers at concentrations similar to those evoked by recombinant protein vaccines (1157, 1163).
Studies in ferrets and non-human primates demonstrated that IWV vaccines can offer protection against infection due to nAb and SARS-CoV-1-specific T cell responses (1164).
However, several attempts to develop IWV vaccines against both SARS-CoV-1 and MERS-CoV were hindered by incidences of vaccine-associated disease enhancement (VADE) in preclinical studies (1165).
In one example of a study in macaques, an inactivated SARS-CoV-1 vaccine induced even more severe lung damage than the virus due to an enhanced immune reaction (1166).
Independent studies in mice also demonstrated evidence of lung immunopathology due to VADE in response to MERS-CoV IWV vaccination (1167, 1168).
The exact mechanisms responsible for VADE remain elusive due to the specificity of the virus-host interactions involved, but VADE is the subject of investigation in preclinical SARS-CoV-2 vaccine studies to ensure the safety of any potential vaccines that may reach phase I trials and beyond (1165).
6.5.1 Application to COVID-19
Table 2: Inactivated whole-virus vaccines approved in at least one country (1169) as of May 3, 2023 (1115).
Vaccine
Company
Covaxin
Bharat Biotech
KoviVac
Chumakov Center
Turkovac
Health Institutes of Turkey
FAKHRAVAC (MIVAC)
Organization of Defensive Innovation and Research
QazVac
Research Institute for Biological Safety Problems (RIBSP)
KCONVAC
Shenzhen Kangtai Biological Products Co
COVIran Barekat
Shifa Pharmed Industrial Co
Covilo
Sinopharm (Beijing)
Inactivated (Vero Cells)
Sinopharm (Wuhan)
CoronaVac
Sinovac
VLA2001
Valneva
Several whole-virus vaccines have been developed against COVID-19 and are available in countries around the world (Table 2).
As of May 31, 2023, 10 vaccines developed with IWV technology are being distributed in 120 countries (Figure 6).
Evidence about the value of these vaccines to combat SARS-CoV-2 is available not only from clinical trials, but also from their roll-out following approval.
Here, a major consideration has been that vaccines often lose efficacy as mutations accumulate in the epitopes of the circulating virus; IWV vaccines may be particularly affected in such cases (1127).
This loss of specificity over time is likely to be influenced by the evolution of the virus, and specifically by the rate of evolution in the region of the genome that codes for the antigenic spike protein.
Here we review three vaccine development programs and their successes in a real-world setting.
Figure 6:Worldwide availability of vaccines developed using inactivated whole viruses.
This figure reflects the number of vaccines based on whole inactivated virus technology that were available in each country as of May 31, 2023.
These data are retrieved from Our World in Data (532) and plotted using geopandas (1170).
The color scale is based on the number of vaccines of this type included in the OWID dataset as a whole, not the maximum observed in a single country.
See https://greenelab.github.io/covid19-review/ for the most recent version of this figure, which is updated daily.
6.5.1.1 Sinovac’s CoronaVac
One IWV vaccine, CoronaVac, was developed by Beijing-based biopharmaceutical company Sinovac.
The developers inactivated a SARS-CoV-2 strain collected in China with β-propiolactone and propagated it using Vero cells (1133).
The vaccine is coupled with an aluminum adjuvant to increase immunogenicity (1133).
Administration follows a prime-boost regimen using a 0.5 ml dose containing 3 μg of inactivated SARS-CoV-2 virus per dose (1171).
In phase I and II clinical trials, CoronaVac elicited a strong immunogenic response in animal models and the development of nAbs in human participants (1172–1174).
The phase I/II clinical trials were conducted in adults 18-59 years old (1174) and adults over 60 years old (1172) in China.
Safety analysis of the CoronaVac vaccine during the phase II trial revealed that most adverse reactions were either mild (grade 1) or moderate (grade 2) in severity (1174).
In adults aged 18 to 59 years receiving a variety of dosage schedules, site injection pain was consistently the most common symptom reported (1174).
In older adults, the most common local and systemic reactions were pain at the injection site (9%) and fever (3%), respectively (1172).
As of December 2022, a total of 17 CoronaVac trials had been registered in a variety of countries including the Philippines and Hong Kong (1175).
Two of the earliest phase III trials to produce results examined a two-dose regimen of CoronaVac following a 14-day prime boost regimen (1176, 1177).
These trials were conducted in Turkey (1177) and Chile (1176) and enrolled participants over an identical period from September 2020 and January 2021.
The Chilean trial, which reported interim results regarding safety and immunogenicity, identified specific IgG nAbs against the S1 RBD and a robust IFN-γ secreting T cell response was induced via immunization with CoronaVac (1176).
In the Turkish trial, VE was estimated to be 83.5% against symptomatic COVID-19 (1177).
In the safety and immunogenicity study, minimal AEs were reported (1177), and 18.9% of participants in the vaccine arm of the Turkish trial reported AEs compared to 16.9% of participants in the placebo group (1177).
However, 2% (n=7) of Turkish participants aged 18 to 59 reported severe AEs (1172), causing the trial to be halted for investigation (1178).
The investigation determined that these events were unrelated to the vaccine (1172, 1178).
An additional phase III trial was conducted in Brazil between July and December 2020 following a randomized, multicenter, endpoint driven, double-blind, placebo-controlled design and enrolling nearly 10,000 healthcare workers (1179, 1180).
The preprint reporting the results of this study (1180) reports an efficacy of 50.7% against symptomatic COVID-19 and 100% against moderate to severe cases.
A large percentage of participants, 77.1% in the vaccine group and 66.4% in the placebo group, reported AEs, including two deaths, but all of the serious AEs were determined not to be related to the vaccine (1180).
CoronaVac also appears to be suitable for use in immunocompromised patients such as those with autoimmune rheumatic diseases according to phase IV trials (1181), and the vaccine was also well tolerated and induced humoral responses in phase I trials in children aged 3 to 17 years, which will now be examined in phase II and III clinical trials (1182).
Estimates of CoronaVac’s VE have varied across trials.
The 50.7% VE reported from the Brazilian trial was contested by Turkish officials, as at the time the efficacy in the Turkish trial appeared to be 91.25% (1183, 1184).
Ultimately, after multiple announcements, the efficacy debate was settled at over 50% (1183, 1184).
Subsequently, the VE for the Turkish trial was finalized at 83.5% (1177), and a prospective national cohort study in Chile reported an adjusted estimated effectiveness of 66% for the prevention of COVID-19 with an estimated 90% and 87% prevention of hospitalization and death, respectively (1185).
Based on these results, CoronaVac was approved in China, and has now been distributed in 66 countries across Africa, Asia, Europe, North America, and South America, including Brazil, Cambodia, Chile, Colombia, Laos, Malaysia, Mexico, Turkey, Ukraine, and Uruguay (1116, 1186).
As of August 2021, Sinovac had reportedly produced over a billion doses of CoronaVac (1186).
Outside of trials, rare cases of VADE have been reported in association with the CoronaVac vaccine (1187).
In one case study, two male patients both presented with COVID-19 pneumonia following vaccination with CoronaVac (1187).
This study identified the timeline of disease presentation, vaccination, and known COVID-19 exposure in the two patients and suggests that the inflammatory response induced by the vaccine could have caused an asymptomatic case of COVID-19 to present with symptoms (1187).
However, no causal relationship between CoronaVac and COVID-19 symptom onset was evaluated, and the reports are extremely rare.
The effectiveness has also been questioned based on real-world data, such as when concerns were raised about the vaccine’s effectiveness following reports that over 350 doctors became ill with COVID-19 in Indonesia despite being immunized with CoronaVac (1188).
One possible explanation for such outbreaks was the evolution of the virus.
Sera from individuals vaccinated with CoronaVac was found to show reduced neutralizing activity against the Alpha, Beta, and Delta VOC relative to the index strain (1189).
Similarly, a second study of 25 patients in Hong Kong in late 2021 evaluated serum neutralizing activity against the index strain and the Beta VOC, Delta VOC, and two Omicron isolates (1190).
They reported that all individuals were seropositive for nAbs against the index strain, 68% against the Delta variant, and 0% against the Beta VOC and Omicron isolates (1190).
The Beta variant appears to be more resistant to nAbs in sera from individuals immunized with CoronaVac than the Alpha variant or wildtype virus, indicating that emerging variants may be of concern (1191).
Finally, a fourth study examined sera from 180 Thai healthcare workers vaccinated with CoronaVac and reported that neutralizing activity was significantly reduced against the Alpha, Delta, and Beta variants relative to the index strain (1192).
Together, these results suggest that viral evolution is likely to pose a significant challenge to immunity acquired from the CoronaVac vaccine.
Therefore, studies have also evaluated whether booster doses would provide additional protection to individuals vaccinated with CoronaVac.
This strategy is supported by the fact that the antibody response elicited by CoronaVac has been found to wane following the second dose, though it was still detected six months out (1193).
A phase I/II clinical trial of CoronaVac in an elderly cohort (adults 60 years and older) in China determined that by 6 to 8 months following the second dose, nAb titers were detected below the seropositive cutoff (1194).
Data from two phase II trials indicated that nAb response had declined 6 months after the second dose of the primary series, but a booster dose of CoronaVac administered 8 months after the second dose markedly increased geometric mean titers of SARS-CoV-2 nAbs (1195).
However, the reduction of nAbs was ameliorated by a booster dose administered 8 months after the second CoronaVac dose (1195).
Furthermore, Chinese (1196) and Chilean (1197) researchers have opted to investigate options to administer different vaccines (e.g., an mRNA vaccine dose) as a booster dose to individuals who have already received two doses of the IWV vaccine CoronaVac.
Another study determined that using a viral-vectored vaccine (CanSino’s Convidecia) or an mRNA vaccine (Pfizer/BioNTech’s BNT162b2) instead of CoronaVac in a prime-boost vaccination regimen could induce a more robust immune response (1198, 1199).
The WHO now suggests that a booster dose, either homologous or heterologous, can be considered 4 to 6 months after the primary series, especially for high-risk groups (1200).
6.5.1.2 Sinopharm’s Covilo
Two additional IWV vaccine candidates were developed following a similar approach by the state-owned China National Pharmaceutical Group Co., Ltd., more commonly known as Sinopharm CNBG.
One, known as BBIBP-CorV or Covilo, was developed in Beijing using the HB02 variant of SARS-CoV-2.
The other was developed at Sinopharm CNBG’s Wuhan Institute using the WIV04 variant of SARS-CoV-2 (1201).
The viruses were purified, propagated using Vero cells, isolated, and inactivated using β-propiolactone (1201, 1202).
Both vaccines are adjuvanted with aluminum hydroxide (1201, 1202).
Here, we focus on Covilo.
Preclinical studies indicated that Covilo induced sufficient nAb titers in mice, and a prime-boost immunization scheme of 2 μg/dose was sufficient to protect rhesus macaques from disease (1202).
In phase II trials, the Covilo vaccine appeared well-tolerated, with 23% of participants in the vaccine condition (482 total participants, 3:1, vaccine:placebo) reporting at least one adverse reaction characterized as mild to moderate (1203).
No evidence of VADE was detected using this vaccine in phase II data (1204).
In phase III trials conducted between July and December 2020, Covilo achieved an efficacy of 72.8% and was well tolerated (1205).
However, questions were raised about efficacy when Sinopharm affiliates in the UAE in early December 2020 claimed the vaccine had 86% efficacy, which was later at odds with a Sinopharm Beijing affiliate that stated that Covilo had a 79.34% efficacy later that same month (1206).
Studies have also investigated expected differences in real-world effectiveness of Covilo given the continuing evolution of SARS-CoV-2.
The antibody response elicited by Covilo was found to wane, but still to be detectable, by six months following the second dose (1193).
One study showed that the Alpha variant exhibited very little resistance to neutralization by sera of those immunized with Covilo, but the Beta variant was more resistant to neutralization by almost a factor of 3 (1191).
Another study examined sera from 282 participants and used a surrogate neutralizing assay, a test that generally correlates with nAbs, to determine that Covilo appears to induce nAbs against the binding of the RBD of wild type SARS-CoV-2 and the Alpha, Beta, and Delta variants to ACE2 (1207).
Notably, a preprint reported that antisera (i.e., the antibody-containing component of the sera) from 12 people immunized with Covilo exhibited nAb capacity against the Beta variant (B.1.351), wild type SARS-CoV-2 (NB02), and one of the original variants of SARS-CoV-2 (D614G) (1208).
As with many other vaccines, booster doses are being evaluated to mitigate some of the issues arising from viral evolution.
A study of healthcare workers in China has since indicated that a booster shot of Covilo elevates B cell and T cell responses and increases nAb titers (1209).
In May 2021, the UAE announced it would consider booster shots for all citizens who had been immunized with Covilo, which was shortly followed by a similar announcement in Bahrain, and by August 29, 2021, the UAE mandated booster shots for all residents who had received Covilo (1186).
6.5.1.3 Bharat Biotech’s Covaxin
Another IWV vaccine candidate was developed by Bharat Biotech International Ltd., which is the biggest producer of vaccines globally, in collaboration with the Indian Council of Medical Research (ICMR) National Institute of Virology (NIV).
This candidate is known as Covaxin or BBV152.
Preclinical studies of Covaxin in hamsters (1210) and macaques (1211) indicated that the vaccine induced protective responses deemed sufficient to move forward to human trials.
Phase I (July 2020) and phase I/II (September to October 2020) studies indicated that Covaxin adjuvanted with alum and a Toll-like receptor 7/8 (TLR7/8) agonist was safe and immunogenic (1212, 1213).
These two studies demonstrated that the vaccine induced significant humoral and cell-mediated responses, as assessed by measuring binding (1212) and neutralizing (1212, 1213) antibodies, cytokines (1212, 1213), CD3+, CD4+, and CD8+ T-cells (1212), with some formulations also eliciting Th1-skewed memory T-cell responses (1213).
Only mild to moderate side-effects were reported upon immunization (1212, 1213), and in phase II trials, the Covilo vaccine appeared well-tolerated (1203).
In India, the Covaxin vaccine received emergency authorization on January 3, 2021, but the phase III data was not released until March 3, 2021, and even then it was communicated via press release (1214).
This press release reported 80.6% efficacy in 25,800 participants (1214, 1215), spurring Zimbabwe to follow suit and authorize the use of Covaxin (1216).
A detailed preprint describing the double-blind, randomized, controlled phase III trial that enrolled between November 2020 and January 2021 became available in July 2021 (1217), and the results collected as of May 17, 2021 were published in December 2021 (1218).
Based on a final enrollment of 25,798 people (~1:1 vaccine:placebo), overall VE against symptomatic COVID-19 was estimated at 77.8% and against severe disease and asymptomatic infection was reported as 93.4% and 63.6%, respectively (1218).
The vaccine was also reported to be well tolerated, with fewer severe events occurring in the Covaxin group (0.3%) than in the placebo group (0.5%) (1218).
One case of a serious AE potentially related to the vaccine, immune thrombocytopenic purpura, was reported, although this patient was seropositive for SARS-CoV-2 at the baseline observation point (1218).
As of June 1, 2023, Covaxin was approved for emergency use in 31 countries across Africa, Asia, Europe, and South America, including Guyana, India, Iran, Zimbabwe, Nepal, Mauritius, Mexico, Nepal, Paraguay, and the Philippines (1219).
Like with all vaccines, the continued evolution of SARS-CoV-2 poses a challenge to the effectiveness of Covaxin.
In this case, the phase III clinical trial did evaluate the efficacy of Covaxin in response to variants circulating in mid-to-late 2020 (1218).
In agreement with previous studies demonstrating sera from individuals vaccinated with Covaxin efficiently neutralized the Alpha variant (B.1.1.7) and the Delta variant (B.1.617.2) (1220–1222), the phase III trial reported a 65.2% efficacy against the Delta variant (B.1.617.2) (1218).
Another study reported that sera from individuals immunized with Covaxin produced effective nAbs against the Delta variant and the so-called Delta plus variant (AY.1) (1223).
Indeed, sera obtained from Covaxin boosted individuals (n=13) (1224) and those who were vaccinated with Covaxin but recovered from a breakthrough infection (n=31) also neutralized the Omicron variant (1225).
Therefore, the data suggest that the vaccine does continue to confer protection to VOC.
The authorization of Covaxin has also offered opportunities to monitor how well the clinical trial results translate into a real-world setting.
Additionally, an effort to monitor AEs and COVID-19 cases following vaccine roll-out reported that most side effects were mild and that cases were rare, even though this data would seem to have been collected during the severe wave of COVID-19 brought on by the Delta VOC in India in early 2021; at the same time, the sample sizes were extremely small (1226).
Similarly, larger studies of adults (June to September 2021) (1227) and adolescents (beginning in January 2022) (1228) who received the vaccine outside of a trial setting reported that safety was similar across age groups, with no severe AEs reported in adults and with no serious AEs reported in adolescents, although 0.9% (6 individuals) reported severe AEs.
However, a much lower effectiveness (22-29%) was estimated in a real-world setting during an analysis of cases in healthcare workers from April to May 2021 (1229).
All the same, monitoring of hospitalized COVID-19 patients between April and June 2021 indicated that the vaccines were highly effective against preventing severe illness (1230).
It is not yet clear what level of protection Covaxin offers beyond 6 to 8 months post the second vaccine; consequently, the potential requirement of a booster immunization is being explored (1231).
Furthermore, Bharat Biotech is considering other vaccine regimens such as providing one initial immunization with Covaxin followed by two immunizations with its intranasal vaccine (BBV154) (1232).
U.S.-based Ocugen Inc., a co-development partner of Bharat Biotech, is leading the application for an Emergency Use Authorization (EUA) for Covaxin intended for the U.S. market.
It has been reported that Bharat Biotech will soon release its phase II and III pediatric trial results (1233).
However, the WHO approval of the Covaxin has been delayed (1234), and in April 2022, the WHO suspended procurement of Covaxin due to concerns about deviation from good manufacturing practice in their production facilities (1235, 1236).
All the same, no safety issues had been reported in association with this vaccine, and the suspension was unlikely to affect distribution given that Bharat Biotech had not been supplying doses through this mechanism (1237).
Clinical trials had recommenced in the United States as of May 2022 (1237).
6.5.2 Summary of IWV Vaccine Development
In the past, problems that arose during the manufacturing of IWV vaccines could present safety issues, but oversight of the manufacturing process has helped to improve IWV vaccine safety (1238).
Nevertheless, the departure from norms necessitated by the COVID-19 crisis raised concerns about whether oversight would occur at pre-pandemic standards (1238).
In general, the IWV COVID-19 vaccines have reported very few issues with safety.
Additionally, safety audits have proactively identified concerns, as demonstrated with the WHO’s suspension of Covaxin.
More concern has arisen around the issue of effectiveness due to the reduced neutralizing activity of IWV vaccines against VOC relative to the index strain.
In several cases, estimates of VE have varied widely across different trials of a single vaccine.
Such issues are likely to be exacerbated by spatiotemporal differences in viral evolution, though in the case of the very high estimate generated by the Turkish trial of CoronaVac (1177), the design of the study may have inflated the VE estimate (1239).
Regardless, the authors of the original trial argued that all of the trials suggest a very high efficacy against severe disease (1240), as is the case for all of the IWV vaccines discussed here.
In addition to issues related to the evolution of SARS-CoV-2, it is important to consider the duration of immunity over time.
With IWV vaccines, heterologous vaccine boosters are being considered in many cases.
Today, the WHO has developed recommendations for booster immunization for several whole-virus vaccines.
In some cases (Valneva-VLA2001 (1241), Covaxin (1242), Covilo (1243), Sinopharm-WIBP Inactivated (Vero Cell) (1244)), boosters are recommended only for high-risk and/or high-priority groups (e.g., the immunocompromised and medical professionals, respectively), while for Sinovac’s CoronaVac (1200), they are recommended more broadly.
Studies are also investigating the effects of booster doses in other vaccines (1245–1247), though some are being investigated or deployed primarily as heterologous boosters in populations vaccinated with a different primary series (1246).
As new vaccines are approved by the WHO, more time elapses since many received the primary series, and new variants emerge, booster recommendations are likely to increase.
Therefore, IWV vaccines have played an important role in vaccine access during the initial phase of vaccination against COVID-19, but many IWV vaccinees may receive booster doses developed with emergent vaccine technologies like DNA and mRNA.
In head-to-head comparisons, these types of vaccines were typically found to outperform IWV vaccines (e.g., (1190, 1192, 1250).
At the same time, IWV vaccines are among the easiest to store and transport due to requiring refrigeration only at 2 to 8°C and remaining stable for years at a time (1205).
Therefore, these vaccines are likely to continue to play an important role in vaccine equity and accessibility.
6.6 Subunit Vaccines
Efforts to overcome the limitations of live-virus vaccines led to the development of approaches first to inactivate viruses (circa 1900), leading to IWV vaccines, and then to purifying proteins from viruses cultured in eggs (circa 1920) (1113, 1251).
The purification of proteins then set the stage for the development of subunit vaccines based on the principle that the immune system can be stimulated by introducing one or more proteins or peptides isolated from the virus.
Today, such approaches often use antigens isolated from the surface of the viral particle that are key targets of the immune system (protein subunit vaccines).
Advances in biological engineering have also facilitated the development of approaches like viral-like particle (VLP) vaccines using nanotechnology (1252).
VLPs share the conformation of a virus’s capsid, thereby acting as an antigen, but lack the replication machinery (1253).
Unlike whole-virus vaccines, which introduce the whole virus, subunit vaccines stimulate the immune system by introducing one or more proteins or peptides of the virus that have been isolated.
The main advantage of this platform is that subunit vaccines are considered very safe, as the antigen alone cannot cause an infection (1254).
Both protein subunit and VLP vaccines thus mimic the principle of whole virus vaccines but lack the genetic material required for replication, removing the risk of infection (1255).
Protein subunit vaccines can stimulate antibodies and CD4+ T-cell responses (1253, 1256).
The subunit approach is also favored for its consistency in production.
The components can be designed for a highly targeted immune response to a specific pathogen using synthetic immunogenic particles, allowing the vaccine to be engineered to avoid allergen and reactogenic sequences (1257).
One limitation is that, in the case of protein subunit vaccines, adjuvants are usually required to boost the immune response (1258) (see online Appendix (1148)).
Adjuvants, which are compounds that elicit an immunogenic effect, include alum (aluminum hydroxide), squalene- or saponin-based adjuvants, and Freund’s incomplete/complete adjuvants, although the latter is avoided in human and veterinary medicine due to high toxicity (1257, 1259, 1260).
Protein subunit vaccine development efforts for both SARS-CoV-1 and MERS-CoV explored a variety of immunogens as potential targets.
The search for a potential SARS-CoV-1 vaccine included the development of vaccines based on the full-length or trimeric S protein (1261–1263), those focused on the RBD protein only (1264–1267) or non-RBD S protein fragments (1262, 1268), and those targeting the N and M proteins (1269–1271).
These efforts have been thoroughly reviewed elsewhere (1272).
There have been examples of successful preclinical research including candidate RBD219N-1, a 218-amino-acid residue of the SARS-CoV-1 RBD that, when adjuvanted to aluminum hydroxide, was capable of eliciting a high antibody response of both nAbs and RBD-specific monoclonal antibodies in both pseudovirus and live virus infections of immunized mice (1273).
Similarly to the SARS-CoV-1 vaccine candidates, the MERS-CoV protein subunit vaccine candidates generally target the RBD (1264, 1272, 1274–1277), with some targeting the full length S protein (1278), non-RBD protein fragments such as the SP3 peptide (1279), and the recombinant N-terminal domain (rNTD) (1280).
Other strategies investigating the potential use of the full length S DNA have also been investigated in mice and rhesus macaques, which elicited immune responses (1281), but these responses were not as effective as the combination of S DNA and the S1 subunit protein together (1281, 1282).
No protein subunit vaccine for MERS-CoV has progressed beyond preclinical research to date.
VLPs have been investigated for development of vaccines against MERS and SARS (1283, 1284) including testing in animal models (1285, 1286), but once again, only preclinical data against HCoV has been collected (1287).
However, protein subunit vaccines do play a role in public health and have contributed to vaccination against hepatitis B (1288) and pertussis (1289, 1290) since the 1980s and human papillomavirus since 2006 (1291).
They are likely to continue to contribute to public health for the foreseeable future due to ongoing research in vaccines against influenza, SARS-CoV-2, Epstein-Barr virus, dengue virus, and human papillomavirus among others (1292–1294).
6.6.1 Application to COVID-19
Table 3: Subunit vaccines approved for use in at least one country (1169) as of May 3, 2023 (1115).
Vaccine
Company
Platform
Zifivax
Anhui Zhifei Longcom
protein subunit
Noora vaccine
Bagheiat-allah University of Medical Sciences
protein subunit
Corbevax
Biological E Limited
protein subunit
Abdala
Center for Genetic Engineering and Biotechnology (CIGB)
protein subunit
Soberana 02
Instituto Finlay de Vacunas Cuba
protein subunit
Soberana Plus
Instituto Finlay de Vacunas Cuba
protein subunit
V-01
Livzon Mabpharm Inc
protein subunit
Covifenz
Medicago
VLP
MVC-COV1901
Medigen
protein subunit
Recombinant SARS-CoV-2 Vaccine (CHO Cell)
National Vaccine and Serum Institute
protein subunit
Nuvaxovid
Novavax
protein subunit
IndoVac
PT Bio Farma
protein subunit
Razi Cov Pars
Razi Vaccine and Serum Research Institute
protein subunit
VidPrevtyn Beta
Sanofi/GSK
protein subunit
COVOVAX (Novavax formulation)
Serum Institute of India
protein subunit
SKYCovione
SK Bioscience Co Ltd
protein subunit
TAK-019 (Novavax formulation)
Takeda
protein subunit
SpikoGen
Vaxine/CinnaGen Co.
protein subunit
Aurora-CoV
Vector State Research Center of Virology and Biotechnology
protein subunit
EpiVacCorona
Vector State Research Center of Virology and Biotechnology
protein subunit
The development of subunit vaccines against SARS-CoV-2 is a remarkable achievement given the short period of time since the emergence of SARS-CoV-2 in late 2019, particularly considering these types of vaccines have not played a major role in previous pandemics compared to LAV and IWV vaccines.
More than 20 protein subunit vaccines from companies such as Sanofi/GlaxoSmithKline, Nanogen, and the Serum Institute of India have entered clinical trials for COVID-19 since the beginning of the pandemic (1293), 20 have been approved, and at least 9 are being administered worldwide (1115, 1116) (Table 3).
As of May 31, 2023, protein subunit vaccines are being distributed in at least 42 countries (Figure 7).
Figure 7:Worldwide availability of vaccines developed using protein subunit.
This figure reflects the number of vaccines based on protein subunit technology that were available in each country as of May 31, 2023.
These data are retrieved from Our World in Data (532, 1116) and plotted using geopandas (1170).
The color scale is based on the number of vaccines of this type included in the OWID dataset as a whole, not the maximum observed in a single country.
See https://greenelab.github.io/covid19-review/ for the most recent version of this figure, which is updated daily.
VLP vaccines have not progressed as rapidly.
Programs seeking to develop VLP vaccines have used either the full-length S protein or the RBD of the S protein specifically as an antigen, although some use several different SARS-CoV-2 proteins (1254).
As of May 31, 2023, only one VLP was available in one country (Canada) (1116).
Figure 8:Worldwide availability of vaccines developed with VLPs.
This figure reflects the number of vaccines based on VLP technology that were available in each country as of May 31, 2023.
These data are retrieved from Our World in Data (532) and plotted using geopandas (1170).
The color scale is based on the number of vaccines of this type included in the OWID dataset as a whole, not the maximum observed in a single country.
See https://greenelab.github.io/covid19-review/ for the most recent version of this figure, which is updated daily.
6.6.2 Novavax’s Nuvaxovid
NVX-CoV2373, also known as Nuvaxovid or Covovax (1295), is a protein subunit vaccine for SARS-CoV-2 produced by U.S. company Novavax and partners.
Nuvaxovid is a protein nanoparticle vaccine constructed from a mutated prefusion SARS-CoV-2 spike protein in combination with a specialized saponin-based adjuvant to elicit an immune response against SARS-CoV-2 (1296).
The spike protein is recombinantly expressed in Sf9 insect cells (1297), which have previously been used for several other FDA-approved protein therapeutics (1298), and contains mutations in the furin cleavage site (682-RRAR-685 to 682-QQAQ-685) along with two proline substitutions (K986P and V987P) that stabilize the protein (1299), including improving thermostability (1297).
In preclinical mouse models, Nuvaxovid elicited high anti-spike IgG titers 21 to 28 days post-vaccination that could neutralize the SARS-CoV-2 virus and protect the animals against viral challenge, with particularly strong effects when administered with the proprietary adjuvant Matrix-MTM(1297).
In a phase I/II trial, a two-dose regimen of Nuvaxovid was found to induce anti-spike IgG levels and nAb titers exceeding those observed in convalescent plasma donated by symptomatic patients (1296).
In line with the preclinical studies, the use of Matrix-M adjuvant further increased anti-spike immunoglobulin levels and induced a Th1 response.
In a phase III randomized, observer-blinded, placebo-controlled clinical trial in the U.K., 14,039 participants received two 5-μg doses of Nuvaxovid or placebo administered 21 days apart in a 1:1 ratio from late September to late November 2020 (1300).
The primary endpoint of the trial was the occurrence or absence of PCR-confirmed, symptomatic mild, moderate or severe COVID-19 from 7 days after the second dose onward (1300).
The VE was reported to be 89.7%, with a total of 10 patients developing COVID-19 in the vaccine group versus 96 in the placebo group (1300).
No hospitalizations or deaths were reported in the vaccine group (1300).
An additional phase III randomized, observer-blinded, placebo-controlled trial was conducted in the U.S. and Mexico, enrolling 29,949 participants and administering at least 1 vaccine in a 2:1 ratio from late December 2020 to late February 2021 (1301).
This trial (1301) used the same primary endpoints as the initial phase III trial conducted in the U.K. (1300).
A vaccine efficacy of 90.4% was reported based on 77 cases total, 63 of which occurred in the placebo group (1301).
All moderate to severe cases of COVID-19 occurred in the placebo group (1301).
Hospitalization and death were not evaluated as individual secondary endpoints, but were instead included in the definition of severe COVID-19; all-cause mortality was comparable between the placebo and treatment conditions (1301).
In both trials, the vaccine was found to be well-tolerated (1300, 1302).
Analysis of 2,310 participants in the U.K. trial revealed that solicited AEs were much more common in the vaccine group than the placebo group across both doses, but the rate of unsolicited events was, while still higher in the vaccine group, much more similar (1300).
A small number of severe AEs were reported by vaccine recipients, including one case of myocarditis; however, the myocarditis was determined to be viral myocarditis (1300).
Common AEs were generally considered mild, with low incidences of headache, muscle pain, and fatigue (1301).
In the trial conducted in the U.S. and Mexico, once again, the most common symptoms included headache, fatigue, and pain, as well as malaise (1301).
Here, severe AEs were balanced across the vaccine and placebo groups (1301).
Thus, both trials suggested that the Nuvaxovid vaccine is safe and effective against COVID-19.
However, Novavax experienced significant challenges in preparing Nuvaxovid for distribution.
Prior to the pandemic’s onset, Novavax had sold their manufacturing facilities and reduced their staff dramatically (1303).
As a result, once they began producing Nuvaxovid, they struggled to establish a stable relationship with contractors who could produce the vaccine (1304), especially given the challenge of producing vaccines at scale (1305).
Additionally, Novavax was not able to meet the purity standards laid out by the FDA (1306).
Eventually, the manufacturing issues were resolved (1307), and Nuvaxovid has since been authorized by the WHO (1308) and by political entities, including the United Kingdom (1309), the E.U. (1310), and the U.S. (1311).
These delays obstructed some of the goals of the vaccine development program, which was undertaken with significant investment from the U.S. government through Operation Warp Speed (1306).
Novavax was supposed to provide over a billion doses of Nuvaxovid to countries around the world through the COVID-19 Vaccines Global Access (COVAX) Facility (1306).
However, following the delays, Gavi (which oversees COVAX) terminated the agreement, leading to ongoing legal disagreements between the nonprofit and Novavax as of late 2022 (1307, 1312).
As with other vaccines, the question of how well Nuvaxovid continues to provide protection as SARS-CoV-2 evolves has been raised.
Post hoc analysis in the phase III trial indicated a VE of 86.3% against the Alpha variant (identified based on the presence of the 69–70del polymorphism) and 96.4% against viral specimens lacking the 69-70del polymorphism (1300).
In the second phase III trial (1302), whole-genome sequencing was obtained from 61 of the 77 observed cases, and 79% of infections were identified as a VOC or variant of interest (VOI) known at the time of the study.
Vaccine efficacy against cases caused by VOC, among which the Alpha variant was predominant (88.6%), was reported to be 92.6% (1302).
In late 2020, an analysis of efficacy in South African adults revealed an overall efficacy of 60.1% and a slightly lower efficacy of 50.1% against the Beta variant (B.1.351) in particular (1313).
The company has also initiated the development of new constructs to select candidates that can be used as a booster against new strains and plans to initiate clinical trials for these new constructs in the second quarter of 2021.
An analysis of a booster dose of Nuvaxovid administered six months after the primary series revealed a significant increase in neutralizing activity against VOC including Delta and Omicron (1314).
This trial was conducted at 18 sites across the United States and Australia (1315).
Novavax has also initiated booster trials in the U.K. (1186).
Boosters may be especially important given that Omicron and related variants, in particular, may be associated with significantly reduced efficacy of Nuvaxovid (1316).
Given the apparent need for boosters, interest has also emerged in whether booster doses of Nuvaxovid can be safely administered along with annual flu vaccines.
In a subgroup of approximately 400 patients enrolled from the U.K. phase III trial who received either Nuvaxovid or a placebo at a ratio of 1:1, a concomitant dose of adjuvanted seasonal influenza vaccines (either a trivalent vaccine or a quadrivalent vaccine) was administered (1317).
This study demonstrated that the vaccines could be safely administered together (1317).
While no change to the immune response was noted for the influenza vaccine, a notable reduction of the antibody response elicited by Nuvaxovid was reported, but efficacy was still high at 87.5% (1317).
Novavax has since started phase I/II trials to investigate the administration of its own influenza vaccine, NanoFlu, concomitantly with Nuvaxovid (1318).
The combination appeared to be safe and effective in preclinical studies (1319).
6.6.3 The Cuban Center for Genetic Engineering and Biotechnology’s Abdala Vaccine
Another notable protein subunit vaccine development program came out of Cuba.
Concerned about their ability to access vaccines, especially given the U.S.’s embargo (1320), health officials in this developing country made the decision in March 2020 to undertake vaccine development on their own (1321).
Today, three Cuban protein subunit vaccines have been approved for use: Abdala, which was developed at the Cuban Genetic Engineering and Biotechnology Center and SOBERANA 02 and SOBERANA Plus, which were developed at Cuba’s Finlay Vaccine Institute (Instituto Finlay de Vacunas Cuba) (1321).
Here, we focus on the development of the Abdala vaccine, but SOBERANA 01/02/Plus vaccine development program has also achieved great success and reported VEs of over 90% in the three-dose regimen (1322, 1323).
Abdala, also known as CIGB-66, was developed using yeast as a low-cost alternative to mammalian cell expression systems (e.g., human embryonic kidney cells) to cultivate the recombinant proteins that form the basis of this protein subunit vaccine (1324).
A sequence corresponding to the RBD of the Spike protein in the index strain of SARS-CoV-2 was codon optimized for expression in yeast, and the RBD proteins were then purified and used to inoculate mice, rats, and African green monkeys (1324).
In addition to the proteins, the vaccine candidate included an adjuvant, aluminum hydroxide gel (1324).
Comparing the immunogenicity of the yeast-cultivated proteins to those cultivated in human embryonic kidney cells revealed no significant difference in the immune response (1324).
Based on promising results in laboratory animal testing, Abdala moved to phase I/II trials in human subjects ages 19 to 80, recruiting participants between December 2020 and February 2021 (1325).
The three-dose vaccine elicited no serious AEs across either phase I or II, and the vaccine was found to produce a strong immune response (1325).
In March 2021, phase III trials began (1326), and by June, officials were reporting the VE to be 92.28% (1327, 1328).
This high efficacy estimate, along with the short timeline of data collection, initially elicited skepticism, especially given that the data were not made public (1329).
However, the trials were designed to enroll a large number of participants and were carried out during a wave of infections due to the arrival of variants carrying the D614G mutation in Cuba, which would be expected to allow an expedited timeline for interim analysis (1329).
Based on the reported results, Abdala gained emergency use authorization in Cuba in July 2021 (1330), and by December 2021, cases in Cuba had dropped dramatically (1331).
The results of the phase III trial were posted to medRxiv in September 2022, describing the results of a randomized, placebo-controlled, multicenter, double-blind investigation of the Abdala vaccine candidate in 48,000 participants between March 22 and April 3, 2021 (1332).
The final results evaluated 42 symptomatic cases of COVID-19 among participants in the placebo condition compared to only 11 cases among participants who received the vaccine, yielding the reported VE of 92.28% (1332).
In terms of secondary endpoints, the VE was 91.96% against mild/moderate COVID-19, 94.46% against severe COVID-19, and 100% against critical illness and death (1332).
The vaccine was also found to be very safe, with the overall incidence of AEs only 2.5% in vaccine recipients compared to 1.9% in the placebo recipients (1332).
Therefore, the phase III trial suggests that this vaccine is highly effective and safe.
Evidence from the deployment of the vaccine also suggests it is highly effective.
A retrospective cohort study conducted between May and August 2021 evaluated public health data from over a million people in the city of Havana and found that the real-world effectiveness of the vaccine met or exceeded estimates of VE during the trial, with 98.2% effectiveness against severe disease and 98.7% effectiveness against death observed in fully vaccinated subjects (1333).
Notably, Cuba has vaccinated a high percentage of its population, with 87% of the population vaccinated by January 2022 and 90.3% by the end of December 2022 (1334, 1335).
Therefore, one consideration in interpreting retrospective cohort studies is that the vaccination rate in Cuba is so high that the two cohorts might not be directly comparable.
All the same, the fact that the efficacy and effectiveness of the Abdala vaccine have both been estimated to be over 90% against severe illness suggests that this vaccine is highly effective for mitigating the risk of COVID-19.
As of December 2022, the vaccine had been authorized for distribution in five additional countries, including Mexico and Vietnam, although its evaluation for WHO approval was ongoing (1336, 1337).
However, limited data is available about the Abdala’s vaccine’s robustness to evolutionary changes in SARS-CoV-2.
An in silico analysis identified several potential changes in the epitopes of the Omicron VOC relative to the sequence used in the development of Abdala (1338).
Instead, Cuban health officials have prioritized boosters.
A representative of the Cuban state business group reportedly stated that immunity remains high at six months after the primary course but that some people may be prone to infection (1339), suggesting waning immunity.
The Cuban government authorized boosters in January 2022 in an effort to mitigate the effects of the Omicron variant (1339–1341).
Additional support for the efficacy of Abdala and other Cuban vaccines comes from the fact that Cuba’s COVID-19 death rate has virtually flatlined since fall 2021, with less than 250 deaths reported during the entire year of 2022 in a population of 11.3 million (1342).
Therefore, in addition to developing a vaccine with an estimated VE paralleling that of vaccines developed using cutting-edge nucleic acid technologies (6), Cuba’s vaccine roll out has also been much more successful than in nearly all similarly sized countries.
This remarkable vaccine program underscores the continued importance of established, cost-effective vaccine development strategies (1340) that make it possible for countries that have not traditionally been a leader in biotechnological innovation but have developed a solid vaccine production sector (1343) to develop and produce vaccines that will serve their own population’s needs.
Additionally, Cuba’s vaccines are uniquely accessible to many countries around the world (1340).
6.6.4 Medicago’s Covifenz
The leading example of a VLP approach applied to COVID-19 comes from Covifenz, developed by Canadian company Medicago (1344).
This vaccine was developed using plant-based VLP technology (1345) that the company had been investigating in order to develop a high-throughput quadrivalent VLP platform to provide protection against influenza (1346).
The approach utilizes Nicotiana benthamiana, an Australian relative of the tobacco plant, as an upstream bioreactor (1346, 1347).
Specifically, the S gene from SARS-CoV-2 in its prefusion conformation is inserted into a bacterial vector (Agrobacterium tumefaciens) that then infects the plant cells (1346, 1347).
Expression of the S glycoprotein causes the production of VLPs composed of S trimers anchored in a lipid envelope that accumulate between the plasma membrane and the cell wall of the plant cell (1347).
Because these VLPs do not contain the SARS-CoV-2 genome, they offer similar advantages to whole-virus vaccines while mitigating the risks (1346, 1347).
In the phase I study, 180 Canadian adults ages 18 to 55 years old were administered Covifenz as two doses, 21 days apart, with three different dosages evaluated (1347).
This study reported that when the VLPs were administered with AS03, an oil-in-water emulsion containing α-tocopherol and squalene (1348), as an adjuvant, the vaccine elicited an nAb response that was significantly (approximately 10 times) higher than that in convalescent sera (1347).
The phase III trial examined 24,141 adults assigned to the treatment and control conditions at a 1:1 ratio between March and September of 2021 (1349).
Covifenz was reported to be 71% effective in preventing COVID-19 in the per-protocol analysis (1349).
Efficacy was only slightly lower in the intention-to-treat group at 69%, with the VE for the prevention of moderate-to-severe disease in this group estimated at 78.8% (1349).
Over 24,000 participants were included in the safety analysis, which reported that 92.3% of vaccine recipients reported local AEs compared to 45.5% of placebo recipients, with rates for systemic AEs at 87.3% and 65.0%, respectively (1349).
The adverse effects reported were generally mild to moderate, with the most common adverse effects being injection site pain, headache, myalgia, fatigue, and general discomfort (1349).
Only three patients (two in the vaccine group) reported grade 4 events, all after the second dose (1349).
The vaccine was approved for use in adults ages 18 to 65 by Health Canada in February 2022 (1350).
Plant-based expression systems such as the one used in Covifenz are relatively new (1347) but are likely to offer unparalleled feasibility at scale given the speed and low-cost associated with the platform (1351).
Additionally, the Covifenz vaccine offers the advantage of being stored at 2 to 8°C.
However, the worldwide footprint of Covifenz, and of VLP-based technologies against SARS-CoV-2 broadly, remains small, with only 1 VLP vaccine approved for distribution in 1 countries (Figure 8).
Approval and administration of Covifenz in countries outside of Canada has been limited by concerns at the WHO about ties between Medicago and the tobacco industry (1344, 1352).
While other species of plants have been explored as the upstream bioreactors for plant-derived VLPs, the specific species of tobacco used increased yield dramatically (1353).
In December 2022, tobacco industry investors in Medicago divested, opening new possibilities for the distribution of the vaccine (1354).
As a result of this limited roll-out and given that the phase III results were published only in May 2022, little is known about the real-world performance of Covifenz.
However, it should be noted that the Covifenz trials were conducted in 2021, at a time during which the B.1.617.2 (Delta) and P.1 (Gamma) variants were predominant (1349).
Genomic analysis of 122 out of 176 cases (165 in the per-protocol population) revealed that none of the COVID-19 cases reported were caused by the original Wuhan strain (1349).
Instead, 45.9% of cases were identified as the Delta variant, 43.4% as Gamma, 4.9% as Alpha, and 5.8% as VOIs (1349).
Therefore, Covifenz and Nuvaxovid, despite both being designed based on the index strain, were tested under circumstances where different VOC were dominant, and differences in the Spike proteins of different VOC relative to the index strain could affect vaccine efficacy.
As of late 2022, Covifenz has not been authorized as a booster in Canada (1355), and no studies on booster doses had been released by Medicago (1356).
6.6.5 Subunit Vaccine Summary
Subunit vaccine technology is one of the best-represented platforms among COVID-19 vaccine candidates.
Development programs are underway in many countries around the world, including low- and middle-income countries (1293).
To date, data about the effect of viral evolution on the effectiveness of subunit vaccines has been limited.
Because these vaccines were developed using the Spike protein from the index strain (1297, 1349), a potential concern has been that these vaccines could lose effectiveness against SARS-CoV-2 containing mutations in the Spike protein.
Comparison of studies across vaccines suggests that some VOC, such as Alpha, may have minimal impact on vaccine efficacy/effectiveness (1357).
Additionally, to the extent that data is available such as from the vaccine rollout in Cuba, it suggests that real-world effectiveness remains strong against severe illness and death.
Subunit platforms offer some unique advantages.
Cuba’s successful vaccine development programs highlights the fact that protein subunit vaccines can be developed using low-cost technologies.
Additionally, they are more feasible to store and transport (1358).
Hoping to build on Cuba’s success and the continued lack of vaccine access in many countries, several Latin American nations have begun developing protein subunit vaccines (1359).
The efficacy and effectiveness of these vaccines is also very high, especially for Nuvaxovid, Abdala, and SOBERANA 01/02/Plus, where estimates exceeded 90%.
Unfortunately, there seem to be limited studies directly comparing the immunogenicity of subunit vaccines to nucleic acid vaccines, and comparing efficacies across trials is subject to bias (1360).
All the same, the evidence suggests that some protein subunit vaccines are able to provide extremely strong protection.
Coupled with the reduced barriers to development and transportation relative to most nucleic acid vaccines, it is clear that subunit technologies are important to vaccine access.
6.7 Global Vaccine Status and Distribution
The unprecedented deployment of COVID-19 vaccines in under a year from the identification of SARS-CoV-2 led to a new challenge: the formation of rapid global vaccine production and distribution plans.
The development of vaccines is costly and complicated, but vaccine distribution can be just as challenging.
Logistical considerations such as transport, storage, equipment (e.g., syringes), the workforce to administer the vaccines, and a continual supply from the manufacturers to meet global demands all must be accounted for and vary globally due to economic, geographic, and sociopolitical reasons (1361–1363).
As of May 25, 2023, at least 13.0 billion vaccine doses had been administered in at least 223 countries worldwide using 28 different vaccines (532).>
The daily global vaccination rate at this time was 8.0 per million.
However, the distribution of these doses is not uniform around the globe.
Latin America leads world vaccination rates with at least 82% of individuals in this region receiving one vaccine dose followed by the U.S. and Canada (81%), Asia-Pacific (81%), Europe (70%), the Middle East (58%), and Africa with only 33% as of November 2022 (1364).
It is estimated that only ~25% of individuals in low- and middle-income countries have received one vaccine dose (1116, 1365).
Vaccine production and distribution varies from region to region and seems to depend on the availability of the vaccines and potentially a country’s resources and wealth (1366).
One effort to reduce these disparities is COVAX, a multilateral initiative as part of the Access to COVID-19 Tools (ACT) Accelerator coordinated by the WHO, Gavi, the Vaccine Alliance, and the Coalition for Epidemic Preparedness Innovations (CEPI), the latter two of which are supported by the Bill and Melinda Gates Foundation.
Their intention is to accelerate the development of COVID-19 vaccines, diagnostics, and therapeutics and to ensure the equitable distribution of vaccines to low- and middle-income countries (1367, 1368).
COVAX invested in several vaccine programs to ensure they would have access to successful vaccine candidates (1369).
However, the initiative has been less successful than was initially hoped due to less participation from high-income countries than was required for COVAX to meet its goals (1370).
Additionally, the vaccine technologies available differ widely around the globe.
As we review elsewhere (6), wealthier nations have invested heavily in mRNA and DNA vaccines.
In contrast, as we describe above, many countries outside of Europe and North America have developed highly effective vaccines using more traditional approaches.
There is a clear relationship between a country’s gross domestic product (GDP) and its access to these cutting-edge types of vaccines (Figure 9).
Whole-virus and subunit vaccine development programs are responsible for a much higher percentage of the vaccinated populous in lower-income countries.
Therefore, vaccine development programs that utilized established vaccine technologies have played a critical role in providing protection against SARS-CoV at the global level.
Figure 9:Vaccine Distribution across Platform Types as a Function of GDP.
The total number of doses of the original formulation of each vaccine that were distributed within each country as of May 31, 2023, by platform type, is shown as a function of GDP.
These data are retrieved from Our World in Data (532, 1116) and plotted using the Python package plotnine (1371).
Lines show a general trend in the data and are drawn using geom_smooth (1372).
The list of countries included in the dataset is available from OWID (1373).
See https://greenelab.github.io/covid19-review/ for the most recent version of this figure, which is updated daily.
Axes are not scaled per capita because both variables are modulated by population size.
When vaccines first became available, the wealthy nations of North America and Europe secured most of the limited COVID-19 vaccine stocks (1374).
Throughout 2021, low- and middle-income countries faced steep competition with high-income countries for vaccines, and the rates of vaccination reflected this unequal distribution (1375).
While the wealthiest countries in these regions could compete with each other for vaccines independent of programs such as COVAX (1375), other countries in these regions have faced challenges in acquiring vaccines developed by the world’s wealthiest nations.
Fortunately, while mRNA and DNA vaccine development programs are not widespread, vaccine development using whole-virus and subunit technologies has been undertaken worldwide.
China and India, in particular, have developed several vaccines that are now widely available in these densely populated countries (see online Appendix (1148)).
Still, many nations, especially in Africa, are reliant on the COVAX Facility, who have promised 600 million doses to the continent (1376).
The COVAX plan seeks to ensure that all participating countries would be allocated vaccines in proportion to their population sizes.
Once each country has received vaccine doses to account for 20% of their population, the country’s risk profile will determine its place in subsequent phases of vaccine distribution.
However, several limitations of this framework exist, including that the COVAX scheme seems to go against the WHO’s own ethical principles of human well-being, equal respect, and global equity and that other frameworks might have been more suitable (1377).
Furthermore, COVAX is supposed to allow poorer countries access to affordable vaccines, but the vaccines are driven by publicly traded companies that are required to make a profit (1366).
In any case, COVAX provides access to COVID-19 vaccines that may otherwise have been difficult for some countries to obtain.
COVAX aimed to distribute 2 billion vaccine doses globally by the end of 2021 (1378).
According to Gavi, as of January 2022, COVAX had distributed over 1 billion vaccines to 144 participants of the program (1379), short of its target but still a major global achievement.
It is envisaged that COVAX may also receive additional donations of doses from Western nations who purchased surplus vaccines in the race to vaccinate their populations, which will be a welcome boost to the vaccination programs of low- and middle-income countries (1380).
In general, deciding on the prioritization and allocation of the COVID-19 vaccines is also a challenging task due to ethical and operational considerations.
Various frameworks, models, and methods have been proposed to tackle these issues with many countries, regions, or U.S. states devising their own distribution and administration plans (1381–1385).
The majority of the distribution plans prioritized offering vaccines to key workers such as health care workers and those who are clinically vulnerable, such as the elderly, the immunocompromised, and individuals with comorbidities, before targeting the rest of the population, who are less likely to experience severe outcomes from COVID-19 (1386).
The availability of vaccines developed in a variety of countries using a variety of platforms is likely to work in favor of worldwide vaccine access.
The initiative by Texas Children’s Hospital and Baylor College of Medicine to develop Corbevax, a patent-free COVID-19 protein subunit vaccine, is an important step towards vaccine equity because the manufacturing specifications can be shared globally.
Corbevax can be produced at low cost using existing technology and is now licensed to Biological E. Limited (BioE), an Indian company specializing in low-cost vaccine production (1387).
The vaccine has been approved for distribution in India and Botswana (1388).
Logistical challenges and geographical barriers also dictate the availability of certain vaccines.
Many countries have had poor availability of ultra-low temperature freezers, leading to challenges of distribution for vaccines such as mRNA vaccines that require storage at very low temperatures (1389–1391).
Furthermore, ancillary supplies such as vaccine containers, diluents for frozen or lyophilized vaccines, disinfecting wipes, bandages, needles, syringes, sharps and biological waste disposal containers are also required, which may not be readily available in geographically isolated locations and can be bulky and expensive to ship (1389).
While some of these challenges in vaccine rollout in low- and middle-income countries are being addressed through COVAX (1392), many issues persist worldwide (1393–1395).
COVAX also failed to distribute its promised two billion vaccine doses on time due to multiple complications (1396).
Another major challenge to global vaccine distribution is vaccine hesitancy, which the WHO has designated as a leading global health threat (1397).
Polling in the U.S. in January 2021 suggested that 20% of individuals were reluctant to receive a vaccine at that time, with a further 31% expressing some hesitancy to a lesser extent (1398, 1399).
A survey of 8,243 long-term healthcare workers in November 2020 (Indiana, USA) reported that only 69% of respondents would ever consider receiving an FDA-approved vaccine due to their perceived risk of side effects (70%), health concerns (34%), efficacy (20%), and religious beliefs (12%) (1400).
Notably, almost a third of parents surveyed in the United States in March 2021 expressed concerns about vaccinating their children against COVID-19 (1401).
Indeed, vaccine hesitancy has been reported as a significant barrier to vaccine distribution in countries in North and South America, Europe, Asia, and Africa (1402–1406).
Various factors have been associated with increased vaccine hesitancy including access to compelling misinformation via social media (1407, 1408), religious and conservative political beliefs (1409–1412), and safety and efficacy concerns (1401), to highlight a few.
Many of the concerns regarding safety and efficacy have focused on the novel mRNA technologies due to the perceived speed of their development and expedited clinical trial process (1413); however, general vaccine hesitancy relating to traditional vaccine platforms existed long before the pandemic and the distribution of the novel mRNA vaccines (1414, 1415).
While in the United States, it was hoped that Novavax’s Nuvaxovid would appeal to the vaccine hesitant (1416, 1417), but this protein subunit vaccine has not led to the uptake hoped (1418, 1419).
Overall, the vaccine landscape remains heterogeneous even as the pandemic nears its third year, with certain vaccines much more accessible in high-income countries than in low- and middle-income countries.
The vaccines described in this manuscript, which were developed using well-established technologies, have played a crucial role in improving the feasibility and accessibility of vaccination programs worldwide.
While the novel technologies have received the bulk of public attention in countries like the U.S., these more traditional vaccine platforms also provide safe and highly effective protection against SARS-CoV-2.
Although companies developing cutting-edge technologies, namely Moderna and Pfizer/BioNTech, reported very high efficacies greater than 90% in their clinical trials (6), the efficacies identified in whole-virus and subunit trials have also been very high.
While the clinical trial efficacy estimates for IWV and subunit have been lower, some of these trials have also reported efficacies over 80% (e.g., Novavax’s Nuvaxovid with 89.7% (1300) or Sinovac’s CoronaVac with 83.5% (1177)).
Variation among studies investigating the efficacy of these vaccines, especially CoronaVac, clearly indicate that clinical trials of the same vaccine might not identify the same efficacy, depending on conditions such as the specific variants circulating in a clinical trial population during the trial period.
Additionally, there are many cohort- and population-level characteristics that can introduce bias within and between clinical trials (1360, 1420), and the extent to which these different factors are present may influence trial outcomes.
While head-to-head comparisons of VE across different studies may therefore not be appropriate, the results make it clear that effective vaccines have been developed with a wide variety of technologies.
The vaccines discussed here, which took advantage of well-established approaches, have proven to be especially valuable in pursuing vaccine equity.
6.8 Conclusions
Much attention has focused on the most novel vaccine technologies that have been deployed against SARS-CoV-2, but the established vaccine platforms discussed here have all made a significant impact on human health during the twentieth century and in some cases even earlier.
The COVID-19 pandemic has demonstrated new potential in these established technologies.
In the early 2000s, these technologies were explored for managing SARS-CoV-1 (1421, 1422), but the epidemic was controlled before those efforts came to fruition (1423).
Similarly, these technologies were explored for MERS-CoV, but outbreaks were sporadic and difficult to predict, making vaccine testing and the development of a vaccination strategy difficult (1424).
However, in the COVID-19 pandemic, most of these technologies have been used to accelerate vaccine development programs worldwide.
Therefore, they are also offering the opportunity to respond quickly to an emergent pathogen.
While these tried-and-true technologies do not always produce vaccines with the highly desirable VE reported in mRNA clinical trials (which exceeded 90%), the efficacies are still very high, and these vaccines are extremely effective at preventing severe illness and death.
Furthermore, some vaccine development programs using established technologies, especially protein subunit vaccines, have seen remarkably high VE and vaccine effectiveness.
Some protein subunit vaccine phase III trials generated VE estimates of over 90%, comparable to those in the mRNA vaccine trials.
Additionally, in some cases, such as Cuba’s highly successful vaccine development program, these vaccines have been developed by and for low- and middle-income nations.
As a result, the greater accessibility and stability of these vaccines makes them extremely valuable for the global effort to mitigate the loss of life from SARS-CoV-2.
The outcomes of the response to COVID-19 suggests that these established vaccine technologies may continue to play an important role in tackling future viral threats.
7 Appendix: Additional Information about Established Vaccine Platforms for COVID-19
7.1 Sinovac’s CoronaVac
The CoronaVac vaccine was developed by Sinovac, a Beijing-based biopharmaceutical company.
The vaccine uses an inactivate whole virus with the addition of an aluminum adjuvant (1425).
Pre-clinical trials were performed using BALB/c mice and rhesus macaques (1173).
The SARS-CoV-2 strains used in this trial isolated from 11 hospitalized patients (5 from China, 3 from Italy, 1 from the United Kingdom (U.K.), 1 from Spain, 1 from Switzerland).
A phylogenetic analysis demonstrated that the strains were representative of the variants circulating at the time.
One of the strains from China, CN2, was used as the inactivated and purified virus while the other 10 strains were used to challenge.
CN2 was grown in Vero cells.
The immunogenicity of the vaccine candidate was evaluated with an ELISA assay.
Ten mice were injected with the vaccine on day 0 and day 7 with varying doses (0, 1.5, 3, or 6 μg), and 10 mice were treated with physiological saline as the control.
IgG developed in the serum of all vaccinated mice.
Using the same setup, immunogenicity was also assessed in macaques.
Four macaques were assigned to each of four groups: treatment with 3 μg at day 0, 7, and 14, treatment with a high dose of 6 μg at day 0, 7, and 14, administration of a placebo vaccine, and administration of only the adjuvant.
All vaccinated macaques induced IgGs and neutralizing antibodies.
After challenge with SARS-CoV-2 strain CN1, vaccinated macaques were protected compared to control macaques (placebo or adjuvant only) based on histology and viral loads collected from different regions of the lung.
A single center, randomized, double-blind, placebo-controlled phase I/II trial was conducted in April 2020i in adults 18-59 years old.
Patients in this study were recruited from the community in Suining County of Jiangsu province, China.
For the phase I trial, 144 (of 185 screened) participants were enrolled, with 72 enrolled in the 14-day interval cohort (i.e., treated on day 0 and day 14) and 72 in the 28-day interval cohort.
This group of 72 participants was split into 2 blocks for a low-dose (3 μg) and high-dose (6 μg) vaccine.
Within each block, participants were randomly assigned vaccination with CoronaVac or placebo (aluminum diluent without the virus) at a 2:1 ratio.
Both the vaccine and placebo were prepared in a Good Manufacturing Practice-accredited facility of Sinovac Life Sciences (Beijing, China).
The phase II trial followed the same organization of participants, this time using 300 enrolled participants in the 14-day and another 300 enrolled in the 28-day groups.
One change of note was that the vaccine was produced using a highly automated bioreactor (ReadyToProcess WAVE 25, GE, Umeå, Sweden) to increase vaccine production capacity.
This change resulted in a higher intact spike protein content.
The authors of this study were not aware of this antigen-level difference between the vaccine batches for the phase I/II when the ethical approval for the trials occurred.
To assess adverse responses, participants were asked to record any events up to 7 days post-treatment.
The reported adverse events were graded according to the China National Medical Products Administration guidelines.
In the phase I trial, the overall incidence of adverse reactions was 29-38% of patients in the 0 to 14 day group and 13-17% in the 0 to 28 day vaccination group.
The most common symptom was pain at the injection site, which was reported by 17-21% of patients in the 0 to 14 day cohort and 13% in the 0 to 28 day cohort.
Most adverse reactions were mild (grade 1), with patients recovering within 48 hours.
A single case of acute hypersensitivity with manifestation of urticaria 48 hours following the first dose of study drug was reported in the 6 μg group
Both the 14-day and 28-day cohorts had a strong neutralizing antibody (Ab) response.
The neutralizing Ab response was measured using a micro cytopathogenic effect assay, which assesses the minimum dilution of neutralizing Ab to be 50% protective against structural changes in host cells in response to viral infection (1426).
Additionally IgG antibody titers against the receptor binding domain were also measured using ELISA.
Another phase I/II study was performed with older patients (over 60 years of age) (1172).
The study conducted a single-center, randomized, double-blind, placebo-controlled trial.
The phase I trial looked at dose escalation using 3 dosages: 1.5, 3, and 6 μg.
The mean age of participants was 65.8 years (standard deviation = 4.8).
Of 95 screened participants, 72 were enrolled.
These 72 participants were split into low (3 μg) and high (6 μg) dose groups.
Within each group, 24 participants received the treatment and 12 the placebo.
A neutralizing antibody response against live SARS-CoV-2 was detected compared to baseline using the same micro cytopathogenic effect assay.
This response was similar across the two dose concentrations.
Additionally, they did not observe a difference in response between age groups (60–64 years, 65–69 years, and ≥70 years).
In phase II the mean age was 66.6 years (standard deviation = 4.7).
499 participants were screened and 350 were enrolled.
300 were evenly split into 1.5, 3, and 6 μg dose groups, and the remaining 50 were assigned to the placebo group.
Again, they found a neutralizing antibody response in phase II.
There wasn’t a significant different between the response to 3 μg versus 6 μg, but the response in both these conditions was higher than in the 1.5 μg condition.
Participants were required to record adverse reaction events within the first 7 days after each dose.
The safety results were combined across phase I and II.
All adverse reactions were either mild (grade 1) or moderate (grade 2) in severity.
The most common symptom was pain at the injection site (9%) and fever (3%).
2% (7 participants) reported severe adverse events (4 from the 1.5 μg group, 1 from the 3 μg group, 2 from the 6 μg group), though these were found to be unrelated to the vaccine.
Overall, the results from the pre-clinical and phase I/II clinical trials were promising, especially the fact that the immune response was consistent in older adults (> 60 years).
7.2 Sinopharm’s Clinical Trials of Two Vaccine Candidates
Sinopharm Wuhan Institute developed their SARS-CoV-2 inactivated vaccine using the WIV04 strain isolated from a patient at the Jinyintan Hospital in Wuhan, China (1201).
This vaccine is administered intramuscularly using 5 μg of virus per dose.
Preclinical data providing supporting evidence for the use of this vaccine is not available publicly.
Despite the lack of publicly available preclinical results, Sinopharm Wuhan Institute initiated phase I/II trials, which reported on varying dosing and prime-boost regimens.
A combined phase I/II RCT of Sinopharm’s BBIBP-CorV, also known as Covilo, followed (1203).
In phase I, 192 participants were randomized with varying doses of 2 μg, 4 μg, or 8 μg/dose or a placebo, and they received the same as a second dose 28 days later.
Approximately 29% of participants reported at least 1 adverse event, most commonly fever, and neutralizing antibody titers were reported for all doses.
In the phase II trial, 482 participants were enrolled (3:1, vaccine:placebo).
Participants in the vaccine condition received either a single 8 μg dose or a double immunization of a 4 μg/dose that was administered 14, 21, or 28 days post the prime dose.
Participants in the placebo condition received the placebo on one of the same four schedules.
The vaccine appeared well-tolerated, with 23% reporting at least one adverse reaction characterized as mild to moderate.
It was reported that all participants had a humoral immune response to the vaccines by day 42 but that the double immunization dosing regimen of 4 μg/dose achieved higher neutralizing antibody titers than a single dose of 8 μg and that the highest response was seen in the double-immunization regimen when at least 21 days separated the two doses (1203).
Similar findings were reported in another phase I/II trial published by the same authors (1205).
For the other vaccine, nAbs were detected in all groups 14 days after the final dose in the phase I part of the trial (1204), with similar findings reported in interim phase II data (1204).
7.3 Novavax’s Nuvaxovid (NVX-CoV2373)
Novavax’s Nuvaxovid is a particularly appealing candidate because the improved stability caused by the proline substitutions is particularly critical to facilitating global distribution, particularly to regions where local refrigerator/freezer capacities are limited.
Importantly, these amino acid substitutions did not affect the ability of the spike protein to bind the hACE2 receptor (the target receptor of SARS-CoV-2 spike protein).
The Novavax-CoV2373 vaccine candidate uses a proprietary, saponin-based Matrix-MTM adjuvant that contains two different 40nm-sized particles formed by formulating purified saponin with cholesterol and phospholipids (1427).
In preclinical models, the use of the Matrix-M adjuvant potentiated the cellular and humoral immune responses to influenza vaccines (1427–1430).
Importantly, Matrix-M adjuvant-containing vaccines have shown acceptable safety profiles in human clinical trials (1431).
Novavax-CoV2373 induced a multifunctional CD4/CD8 T-cell responses and generate high frequencies of follicular helper T-cells and B-cell germinal centers after vaccination.
These findings were subsequently evaluated in a baboon primate model, in which Novavax-CoV2373 also elicited high antibody titers against the SARS-CoV-2 spike protein, as well as an antigen specific T-cell response.
Based on this data Novavax initiated a phase I/II clinical trial to evaluate the safety and immunogenicity of Novavax-CoV2373 with Matrix-M (1296, 1315).
The phase I/II trial was a randomized, placebo-controlled study with 131 healthy adult participants in 5 treatment arms (1296).
Participants that received the recombinant SARS-CoV-2 vaccine with or without the Matrix-M adjuvant got two injections, 21 days apart.
Primary outcomes that were assessed include reactogenicity, lab-values (serum chemistry and hematology), and anti-spike IgG levels.
Secondary outcomes measured included virus neutralization, T-cell responses, and unsolicited adverse events.
The authors reported that no serious treatment-related adverse events occurred in any of the treatment arms.
Reactogenicity was mostly absent and of short duration.
The two-dose vaccine regimen induced anti-spike IgG levels and neutralizing antibody-titers exceeding those in the convalescent plasma of symptomatic patients.
The outcomes of this trial suggest that Novavax-CoV2373 has an acceptable safety profile and is able to induce a strong immune response with high neutralizing antibody titers.
The phase II component of the trial was designed to identify the dose regimen for the next clinical trial stage (1432).
Both younger (18-59 years) and older patients (60-84 years) were randomly assigned to receive either 5 μg or 25 μg Novavax-CoV2373 or a sodium-chloride placebo in two doses, 21 days apart.
In line with the phase I data, reactogenicity remained mild to moderate, with no more than 1% of participants in any group reporting grade 3 AEs, and of short duration.
Both dose levels were able to induce high anti-spike IgG titers as well as neutralizing antibody responses after the second dose.
Based on both safety and efficacy data, the 5 μg dosing regimen was selected as the optimal dose regimen for the phase III trial.
Novavax announced an efficacy of 89.3% based on their phase III trial in the U.K. and South Africa (1300, 1433, 1434).
This trial included over 15,000 participants in the U.K. and 4,000 participants in South Africa.
The primary endpoint of the trial was the occurrence or absence of PCR-confirmed, symptomatic mild, moderate or severe COVID-19 from 7 days after the second dose onward.
In the first interim analysis (U.K.), 56 cases of COVID-19 were observed in the placebo group compared to 6 cases in the treatment group.
Importantly, the vaccine candidate also shows significant clinical efficacy against the prevalent U.K. and South African variants.
7.4 Protein Subunit Vaccine Development Programs Prior to SARS-CoV-2
Earlier studies examined the immunogenicity of a SARS-CoV-1 RBD fused with IgG1 Fc.
This recombinant fusion protein could induce a robust long-lasting neutralizing antibody and cellular immune response that protected mice from SARS-CoV-1 (1133, 1264, 1267).
While there have been other potential protein subunit vaccines for SARS-CoV-1 investigated in vivo(1133, 1272), none of these candidates have successfully completed clinical trials, more than likely due to the fact that the SARS-CoV-1 epidemic mostly ended by May 2004, and there was thus less of a demand or funding for SARS-CoV-1 vaccine research.
Similar vaccine candidates have emerged that target the RBD found in the S1 subunit of the trimeric MERS-CoV S protein, which binds to dipeptidyl-peptidase 4 (DPP4 also known as hCD26), the entry point through which MERS-CoV infects cells (1435–1437).
After initially determining that an RBD subunit candidate (S377-588-Fc) could elicit neutralizing antibodies (1438), a study in mice determined that the administration of three sequential doses of RBD-Fc vaccine coupled with MF59, a squalene immunogenic adjuvant, induced humoral and systemic immunity in mice (1439).
Mice that had been transduced with Ad5-hCD26 and subsequently challenged with MERS-CoV five days later did not show evidence of viral infection in the lungs versus control mice at ten days post vaccination (1439).
Other variations of this vaccine approach include a stable S trimer vaccine whereby proline-substituted variants of S2 can maintain a stable prefusion conformation of the S2 domain (1133).
This approach leads to broad and potent neutralizing antibodies (1133)
7.5 Complementary Approaches to Vaccine Development
A complementary approach to other vaccine development programs that is being investigated explores the potential for vaccines that are not made from the SARS-CoV-2 virus to confer what has been termed trained immunity.
In a recent review (1440), trained immunity was defined as forms of memory that are temporary (e.g., months or years) and reversible.
It is induced by exposure to whole-microorganism vaccines or other microbial stimuli that generates heterologous protective effects.
Trained immunity can be displayed by innate immune cells or innate immune features of other cells, and it is characterized by alterations to immune responsiveness to future immune challenges due to epigenetic and metabolic mechanisms.
These alterations can take the form of either an increased or decreased response to immune challenge by a pathogen.
Trained immunity elicited by non-SARS-CoV-2 whole-microorganism vaccines could potentially improve SARS-CoV-2 susceptibility or severity (1441).
One type of stimulus which research indicates can induce trained immunity is bacillus Calmette-Guerin (BCG) vaccination.
BCG is an attenuated form of bacteria Mycobacterium bovis.
The vaccine is most commonly administered for the prevention of tuberculosis in humans.
Clinical trials in non-SARS-CoV-2-infected adults have been designed to assess whether BCG vaccination could have prophylactic effects against SARS-CoV-2 by reducing susceptibility, preventing infection, or reducing disease severity.
A number of trials are now evaluating the effects of the BCG vaccine or the related vaccine VPM1002 (1151, 1152, 1441–1453).
The ongoing trials are using a number of different approaches.
Some trials enroll healthcare workers, other trials hospitalized elderly adults without immunosuppression who get vaccinated with placebo or BCG at hospital discharge, and yet another set of trials older adults (>50 years) under chronic care for conditions like hypertension and diabetes.
One set of trials, for example, uses time until first infection as the primary study endpoint; more generally, outcomes measured in some of these trials are related to incidence of disease and disease severity or symptoms.
Some analyses have suggested a possible correlation at the country level between the frequency of BCG vaccination (or BCG vaccination policies) and the severity of COVID-19 (1441).
Currently it is unclear whether this correlation has any connection to trained immunity.
Many possible confounding factors are also likely to vary among countries, such as age distribution, detection efficiency, stochastic epidemic dynamic effects, differences in healthcare capacity over time in relation to epidemic dynamics, and these have not been adequately accounted for in current analyses.
It is unclear whether there is an effect of the timing of BCG vaccination, both during an individual’s life cycle and relative to the COVID-19 pandemic.
Additionally, given that severe SARS-CoV-2 may be associated with a dysregulated immune response, it is unclear what alterations to the immune response would be most likely to be protective versus pathogenic (e.g., (149, 1441, 1454, 1455)).
The article (1441) proposes that trained immunity might lead to an earlier and stronger response, which could in turn reduce viremia and the risk of later, detrimental immunopathology.
While trained immunity is an interesting possible avenue to complement vaccine development efforts through the use of an existing vaccine, additional research is required to assess whether the BCG vaccine is likely to confer trained immunity in the case of SARS-CoV-2.
7.6 India and China’s Roles in Vaccine Innovation and Development
The nations of China and India have played a major role as COVID-19 vaccination developers and providers.
Considering India produced approximately 60% of the world’s vaccines prior to the pandemic, it is no surprise that the nation has developed and is developing several COVID-19 vaccine candidates.
In addition to Covaxin, the Bio E subunit vaccine CORBEVAX is being produced by Biological E in collaboration with U.S.-based Dynavax and the Baylor College of Medicine (1456).
These two home-grown vaccines are now approved for adults and children as young as five (CORBEVAX) and six (Covaxin) (1457).
Other vaccines licensed by India were developed elsewhere but produced in India.
For example, Novavax (developed in the United States) has signed an agreement with the Serum Institute of India allowing them to produce up to 2 billion doses a year (1458).
Similarly, many people within India have been vaccinated with the AstraZeneca-University of Oxford vaccine, known as Covishield in India, which is also produced by the Serum Institute of India (1456).
India is also developing vaccines using cutting-edge nucleic-acid-based platforms..
These include ZyCov-D, a DNA vaccine produced by Zydus Cadila, HGCO19 and India’s first mRNA vaccine, produced by Genova and HDT Biotech Corporation (of the U.S.) (1456).
Additionally, in February 2021, Bharat Biotech received approval from Indian officials to commence a phase I study of an intranasal chimpanzee-adenovirus (ChAd) vectored SARS-CoV-2-S vaccine called BBV154 (1459).
In China, the Sinopharm-Beijing Institute vaccine, the Sinopharm-Wuhan Institute of Biological Products vaccine, the Sinovac Biotech (CoronaVac) vaccine, and CanSino Biologics vaccine are the main vaccines being distributed.
Sinovac and Sinopharm aimed to produce 2 billion doses by the end of 2021, and they have distributed vaccines as aid to the Philippines and Pakistan (1460).
In contrast, the Sinopharm-Wuhan vaccine, which has been approved for use in China since February 25, 2021, has been distributed almost exclusively within China, with limited supplies distributed to the United Arab Emirates (1186).
On the same date, the CanSino vaccine was approved for use in China and has been granted emergency use in several other countries (1186).
However, the vaccine approval and distribution processes in China have come under increased scrutiny from other nations.
China was criticized for administering vaccines to thousands of government officials and state-owned businesses in September 2020, prior to the completion of phase III clinical trials (1461).
The behavior of Chinese officials has also come into question due to misinformation campaigns questioning the safety of Western vaccine candidates such as Moderna and Pfizer-BioNTech in a way that is intended to highlight the benefits of their own vaccine candidates (1460).
China in particular took aim at mRNA technologies, but Chinese companies have since developed their own mRNA vaccines targeting the omicron variant, one of which is due to begin trials soon in the UAE (1462).
Furthermore, delays in vaccine distribution have also caused issues, particularly in Turkey where 10 million doses of Sinovac were due to arrive by December 2020, but instead only 3 million were delivered in early January (1460).
Similar delays and shortages of doses promised were reported by officials in the Philippines, Egypt, Morocco, and the United Arab Emirates (1463, 1464).
All the same, Sinovac’s vaccine has since been approved for use in countries around the world (1186).
8 The Coming of Age of Nucleic Acid Vaccines during COVID-19
8.1 Abstract
In the 21st century, several emergent viruses have posed a global threat.
Each pathogen has emphasized the value of rapid and scalable vaccine development programs.
The ongoing SARS-CoV-2 pandemic has made the importance of such efforts especially clear.
New biotechnological advances in vaccinology allow for recent advances that provide only the nucleic acid building blocks of an antigen, eliminating many safety concerns.
During the COVID-19 pandemic, these DNA and RNA vaccines have facilitated the development and deployment of vaccines at an unprecedented pace.
This success was attributable at least in part to broader shifts in scientific research relative to prior epidemics; the genome of SARS-CoV-2 was available as early as January 2020, facilitating global efforts in the development of DNA and RNA vaccines within two weeks of the international community becoming aware of the new viral threat.
Additionally, these technologies that were previously only theoretical are not only safe but also highly efficacious.
Although historically a slow process, the rapid development of vaccines during the COVID-19 crisis reveals a major shift in vaccine technologies.
Here, we provide historical context for the emergence of these paradigm-shifting vaccines.
We describe several DNA and RNA vaccines and in terms of their efficacy, safety, and approval status.
We also discuss patterns in worldwide distribution.
The advances made since early 2020 provide an exceptional illustration of how rapidly vaccine development technology has advanced in the last two decades in particular and suggest a new era in vaccines against emerging pathogens.
8.2 Importance
The SARS-CoV-2 pandemic has caused untold damage globally, presenting unusual demands on but also unique opportunities for vaccine development.
The development, production, and distribution of vaccines is imperative to saving lives, preventing severe illness, and reducing the economic and social burdens caused by the COVID-19 pandemic.
Although vaccine technologies that provide the DNA or RNA sequence of an antigen had never previously been approved for use in humans, they have played a major role in the management of SARS-CoV-2.
In this review we discuss the history of these vaccines and how they have been applied to SARS-CoV-2.
Additionally, given that the evolution of new SARS-CoV-2 variants continues to present a significant challenge in 2022, these vaccines remain an important and evolving tool in the biomedical response to the pandemic.
8.3 Introduction
The SARS-CoV-2 virus emerged at the end of 2019 and soon spread around the world.
In response, the Coalition for Epidemic Preparedness Innovations quickly began coordinating global health agencies and pharmaceutical companies to develop vaccines, as vaccination is one of the primary approaches available to combat the effects of a virus.
Vaccines can bolster the immune response to a virus at both the individual and population levels, thereby reducing fatalities and severe illness and potentially driving a lower rate of infection even for a highly infectious virus like SARS-CoV-2.
However, vaccines have historically required a lengthy development process due to both the experimental and regulatory demands.
As we review in a companion manuscript (5), vaccine technologies prior to the COVID-19 pandemic were largely based on triggering an immune response by introducing a virus or one of its components.
Such vaccines are designed to induce an adaptive immune response without causing the associated viral illness.
Each time a virus emerges that poses a significant global threat, as has happened several times over the past 20 years, the value of a rapid vaccine response is underscored.
With progressive biotechnological developments, this objective has become increasingly tangible.
In the current century, significant advances in vaccine development have largely been built on genomics, as is somewhat unsurprising given the impact of the Genomic Revolution across all biology.
This shift towards nucleic acid-based technologies opens a new frontier in vaccinology, where just the sequence encoding an antigen can be introduced to induce an immune response.
While other platforms can carry some risks related to introducing all or part of a virus (5), nucleic acid-based platforms eliminate these risks entirely.
Additionally, vaccine technologies that could be adjusted for novel viral threats are appealing because this modular approach would mean they could enter trials quickly in response to a new pathogen of concern.
8.4 Honing a 21st Century Response to Emergent Viral Threats
Recently, vaccine technologies have been developed and refined in response to several epidemics that did not reach the level of destruction caused by COVID-19.
Emergent viral threats of the 21st century include severe acute respiratory syndrome (SARS), the H1N1 influenza strain known as swine flu, Middle East respiratory syndrome (MERS), Ebola virus disease, COVID-19, and, most recently, monkeypox, all of which have underscored the importance of a rapid global response to a new infectious virus.
Because the vaccine development process has historically been slow, the use of vaccines to control most of these epidemics was limited.
One of the more successful recent vaccine development programs was for H1N1 influenza.
This program benefited from the strong existing infrastructure for influenza vaccines along with the fact that regulatory agencies had determined that vaccines produced using egg- and cell-based platforms could be licensed under the regulations used for a strain change (1423).
Although a monovalent H1N1 vaccine was not available before the pandemic peaked in the United States of America (U.S.A.) and Europe, it became available soon afterward as a stand-alone vaccine that was eventually incorporated into commercially available seasonal influenza vaccines (1423).
Critiques of the production and distribution of the H1N1 vaccine have stressed the need for alternative development-and-manufacturing platforms that can be readily adapted to new pathogens.
Efforts to develop such approaches had been undertaken prior to the COVID-19 pandemic.
DNA vaccine development efforts began for SARS-CoV-1 but did not proceed past animal testing (1422).
Likewise, the development of viral-vectored Ebola virus vaccines was undertaken, but the pace of vaccine development was behind the spread of the virus from early on (1465).
Although a candidate Ebola vaccine V920 showed promise in preclinical and clinical testing, it did not receive breakthrough therapy designation until the summer of 2016, by which time the Ebola outbreak was winding down (1466).
Therefore, the COVID-19 pandemic has been the first case where vaccines have been available early enough to significantly influence outcomes at the global scale.
The pandemic caused by SARS-CoV-2 has highlighted a confluence of circumstances that positioned vaccine development as a key player in efforts to control the virus and mitigate its damage.
This virus did not follow the same trajectory as other emergent viruses of recent note, such as SARS-CoV-1, MERS-CoV, and Ebola virus, none of which presented a global threat for such a sustained duration (see visualization in (3)).
Spread of the SARS-CoV-2 virus has remained out of control in many parts of the world into 2022, especially with the emergence of novel variants exhibiting increased rates of transmission (1).
While, for a variety of reasons, SARS-CoV-2 was not controlled as rapidly as the viruses underlying prior 21st century epidemics, vaccine development technology had also progressed based on these and other prior viral threats to the point that a rapid international vaccine development response was possible.
8.5 Development of COVID-19 Vaccines using DNA and RNA Platforms
Vaccine development programs for COVID-19 emerged very quickly.
The first administration of a dose of a COVID-19 vaccine to a human trial participant occurred on March 16, 2020 (1467, 1468), marking an extremely rapid response to the emergence of SARS-CoV-2.
Within a few weeks of this first trial launching, at least 78 vaccine development programs were active (1468), and by September 2020, there were over 180 vaccine candidates against SARS-CoV-2 in development (1114).
As of May 3, 2023, 50 SARS-CoV-2 vaccines have been approved world wide and 28 are being administered throughout the world, with 13.0 billion doses administered across 223 countries.
The first critical step towards developing a vaccine against SARS-CoV-2 was characterizing the viral target, which happened extremely early in the COVID-19 outbreak with the sequencing and dissemination of the viral genome in early January 2020 (555) (Figure 10).
This genomic information allowed for an early identification of the sequence of the Spike (S) protein (Figure 10), which is the antigen and induces an immune response (1469, 1470).
Figure 10:Structure of the SARS-CoV-2 virus.
The development of vaccines depends on the immune system recognizing the virus.
Here, the structure of SARS-CoV-2 is represented both in the abstract and against a visualization of the virion.
The abstracted visualization was made using BioRender (1471) using the template “Human Coronavirus Structure” by BioRender (August 2020) (1472).
The microscopy was conducted by the National Institute of Allergy and Infectious Diseases (1473).
During the development process, one measure used to assess whether a vaccine candidate is likely to provide protection is serum neutralizing activity (1119).
This assay evaluates the presence of antibodies that can neutralize, or prevent infection by, the virus in question.
Often, titration is used to determine the extent of neutralization activity.
However, unlike in efforts to develop vaccines for prior viral threats, the duration of the COVID-19 pandemic has made it possible to also test vaccines in phase III trials where the effect of the vaccines on a cohort’s likelihood of contracting SARS-CoV-2 was evaluated.
8.6 Theory and Implementation of Nucleic Acid Vaccines
Biomedical research in the 21st century has been significantly influenced by the genomic revolution.
While traditional methods of vaccine development, such as inactivated whole viruses are still used today (5), vaccine development is no exception.
The shift towards omics-based approaches to vaccine development began to take hold with the meningococcal type B vaccine, which was developed using reverse vaccinology in the early 2010s (1474, 1475).
Under this approach, the genome of a pathogen is screened to identify potential vaccine targets (1475), and pathogens of interest are then expressed in vitro and tested in animal models to determine their immunogenicity (1475).
In this way, the genomic revolution catalyzed a fundamental shift in the development of vaccines.
Such technologies could revolutionize the role of vaccines given their potential to address one of the major limitations of vaccines today and facilitate the design of therapeutic, rather than just prophylactic, vaccines (1476).
Nucleic-acid based approaches share an underlying principle: a vector that delivers the information needed to produce an antigen.
When the host cells manufacture the antigen, it can then trigger an immune response.
The fact that no part of the virus is introduced aside from the genetic code of the antigen means that these vaccines carry no risk of infection.
Such approaches build on subunit vaccination strategies, where a component of a virus (e.g., an antigenic protein) is delivered by the vaccine.
Platforms based on genomic sequencing began to be explored beginning in the 1980s as genetic research became increasingly feasible.
Advances in genetic engineering allowed for gene sequences of specific viral antigens to be grown in vitro(1113).
Studies also demonstrated that model organisms could be induced to construct antigens that would trigger an immune response (1128, 1477, 1478).
These two developments sparked interest in whether it could be possible to identify any or all of the antigens encoded by a virus’s genome and train the immune response to recognize them.
The delivery and presentation of antigens is fundamental to inducing immunity against a virus.
Vaccines that deliver nucleic acids allow the introduction of foreign substances to the body to induce both humoral and cellular immune responses (1479).
Delivering a nucleic acid sequence to host cells allows the host to synthesize an antigen without exposure to a viral threat (1479).
Host-synthesized antigens can activate both humoral and cellular immunity (1479), as they can be presented in complex with major histocompatibility complex (MHC) I and II, which can activate either T- or B-cells (1479).
In contrast, prior approaches activated only MHC II (1478).
Because these vaccines encode specific proteins, providing many of the benefits of a protein subunit vaccine, they do not carry any risk of DNA being live, replicating, or spreading, and their manufacturing process lends itself to scalability (1479).
Here, opportunities can be framed in terms of the central dogma of genetics: instead of directly providing the proteins from the infectious agents, vaccines developers are exploring the potential for the delivery of DNA or RNA to induce the cell to produce proteins from the virus that in turn induce a host immune response.
8.7 Cross-Platform Considerations in Vaccine Development
Certain design decisions are relevant to vaccine development across multiple platforms.
One applies to the platforms that deliver the antigen, which in the case of SARS-CoV-2 vaccines is the S protein.
The prefusion conformation of the S protein, which is the structure before the virus fuses to the host cell membrane, is metastable (1480), and the release of energy during membrane fusion drives this process forward following destabilization (12, 1481).
Due to the significant conformational changes that occur during membrane fusion (25, 1482, 1483), S protein immunogens that are stabilized in the prefusion conformation are of particular interest, especially because a prefusion stabilized Middle East respiratory syndrome-related coronavirus (MERS-CoV) S antigen was found to elicit an improved antibody response (1278).
Moreover, the prefusion conformation offers an opportunity to target S2, a region of the S protein that accumulates mutations at a slower rate (23, 24, 1278) (see also (1)).
Vaccine developers can stabilize the prefusion conformer by selecting versions of the S protein containing mutations that lock the position (1484).
The immune response to the Spike protein when it is stabilized in this conformation is improved over other S structures (1485).
Thus, vaccines that use this prefusion stabilized conformation are expected to not only offer improved immunogenicity, but also be more resilient to the accumulation of mutations in SARS-CoV-2.
Another cross-platform consideration is the use of adjuvants.
Adjuvants include a variety of molecules or larger microbial-related products that affect the immune system broadly or an immune response of interest.
They can either be comprised of or contain immunostimulants or immunomodulators.
Adjuvants are sometimes included within vaccines in order to enhance the immune response.
Different adjuvants can regulate different types of immune responses, so the type or combination of adjuvants used in a vaccine will depend on both the type of vaccine and concern related to efficacy and safety.
A variety of possible mechanisms for adjuvants have been investigated (1486–1488).
Due to viral evolution, vaccine developers are in an arms race with a pathogen that benefits from mutations that reduce its susceptibility to adaptive immunity.
The evolution of several variants of concern (VOC) presents significant challenges for vaccines developed based on the index strain identified in Wuhan in late 2019.
We discuss these variants in depth elsewhere in the COVID-19 Review Consortium project (1148).
To date, the most significant variants of concern identified are Alpha (2020), Beta (2020), Gamma (2020), Delta (2021), Omicron (2021), and related Omicron subvariants (2022).
The effectiveness or efficacy (i.e., trial or real-world prevention, respectively) of vaccines in the context of these variants is discussed where information is available.
8.8 DNA Vaccine Platforms
DNA vaccine technologies have developed slowly over the past thirty years.
These vaccines introduce a vector containing a DNA sequence that encodes antigen(s) selected to induce a specific immune response (1478).
Early attempts revealed issues with low immunogenicity (1128, 1478, 1489).
Additionally, initial skepticism about the approach suggested that DNA vaccines might bind to the host genome or induce autoimmune disease (1479, 1490), but pre-clinical and clinical studies have consistently disproved this hypothesis and indicated DNA vaccines to be safe (1489).
Another concern, antibiotic resistance introduced during the plasmid selection process, did remain a concern during this initial phase of development (1479), but this issue was resolved through strategic vector design (1491, 1492).
However, for many years, the immunogenicity of DNA vaccines failed to reach expectations (1479).
Several developments during the 2010s led to greater efficacy of DNA vaccines (1479).
However, no DNA vaccines had been approved for use in humans prior to the COVID-19 pandemic (1489, 1493).
As of May 3, 2023, 10 vaccines have been approved worldwide (Table 4).
These vaccines fall into two categories, vaccines that are vectored with a plasmid and those that are vectored with another virus.
Table 4: DNA vaccines approved in at least one country (1169) as of May 3, 2023.
Vaccine
Company
Platform
iNCOVACC
Bharat Biotech
non replicating viral vector
Convidecia
CanSino
non replicating viral vector
Convidecia Air
CanSino
non replicating viral vector
Gam-COVID-Vac
Gamaleya
non replicating viral vector
Sputnik Light
Gamaleya
non replicating viral vector
Sputnik V
Gamaleya
non replicating viral vector
Jcovden
Janssen (Johnson & Johnson)
non replicating viral vector
Vaxzevria
Oxford/AstraZeneca
non replicating viral vector
Covishield (Oxford/ AstraZeneca formulation)
Serum Institute of India
non replicating viral vector
ZyCoV-D
Zydus Cadila
plasmid vectored
8.8.1 Plasmid-Vectored DNA Vaccines
Many DNA vaccines use a plasmid vector-based approach, where the sequence encoding the antigen(s) against which an immune response is sought are cultivated in a plasmid and delivered directly to an appropriate tissue (1494).
Plasmids can also be designed to act as adjuvants by targeting essential regulators of pathways such as the inflammasome or simply just specific cytokines (1490, 1495).
The DNA itself may also stimulate the innate immune response (1478, 1492).
Once the plasmid brings the DNA sequence to an antigen-presenting cell (APC), the host machinery can be used to construct antigen(s) from the transported genetic material, and the body can then synthesize antibodies in response (1479).
The vectors are edited to remove extra sequences (1492).
These types of manufacturing advances have improved the safety and throughput of this platform (1492).
8.8.1.1 Prior Applications
In the 1990s and 2000s, DNA vaccines delivered via plasmids sparked significant scientific interest, leading to a large number of preclinical trials (1479).
Early preclinical trials primarily focused on long-standing disease threats, including viral diseases such as rabies and parasitic diseases such as malaria, and promising results led to phase I testing of the application of this technology to human immunodeficiency virus (HIV), influenza, malaria, and other diseases of concern during this period (1479).
Although they were well-tolerated, these early attempts to develop vaccines were generally not very successful in inducing immunity to the target pathogen, with either limited T-cell or limited neutralizing antibody responses observed (1479).
Early plasmid-vectored DNA vaccine trials targeted HIV and subsequently diseases of worldwide importance such as malaria and hepatitis B (1496).
The concern with these early development projects was immunogenicity, not safety (1496).
Around the turn of the millennium, a hepatitis B vaccine development program demonstrated that these vaccines can induce both antibody and cellular immune response (1497).
Prior to COVID-19, however, plasmid-vectored DNA vaccines had been approved for commercial use only in veterinary populations (1498–1500).
Between 2005 and 2006, several DNA vaccines were developed for non-human animal populations, including against viruses including a rhabdovirus in fish (1501), porcine reproductive and respiratory syndrome virus (1502), and West Nile virus in horses (1503).
Within the past five years, additional plasmid-vectored vaccines for immunization against viruses were developed against a herpesvirus (in mice) (1504) and an alphavirus (in fish) (1505).
8.8.1.2 Applications to COVID-19
Several plasmid-vectored DNA vaccines have been developed against COVID-19 (Table 4).
In fact, the ZyCoV-D vaccines developed by India’s Zydus Cadila is the first plasmid-vectored DNA vaccine to receive approval or to be used in human medicine (1506–1508).
Another plasmid-vectored DNA vaccine, INO-4800 (1509), was developed by Inovio Pharmaceuticals Technology that uses electroporation as an adjuvant.
Electroporation was developed as a solution to the issue of limited immunogenicity by increasing the permeability of cell membranes by delivering electrical pulses (1510).
It has been shown that electroporation can enhance vaccine efficacy by up to 100-fold, as measured by increases in antigen-specific antibody titers (1511).
The temporary formation of pores through electroporation facilitates the successful transportation of macromolecules into cells, allowing cells to robustly take up INO-4800 for the production of an antibody response.
For INO-4800, a plasmid-vectored vaccine is delivered through intradermal injection which is then followed by electroporation with a device known as CELLECTRA® (1512).
The safety of the CELLECTRA® device has been studied for over seven years, and these studies support the further development of electroporation as a safe vaccine delivery method (1510).
These vaccines therefore represent implementations of a new platform technology.
In particular, they offer the advantage of a temperature-stable vaccine, facilitating worldwide administration (1513).
Although an exciting development in DNA vaccines, the cost of vaccine manufacturing and electroporation may make scaling the use of this technology for prophylactic use for the general public difficult.
8.8.1.3 Trial Safety and Immunogenicity
The INO-4800 trials began with a phase I trial evaluating two different doses administered as a two-dose series (1512).
This trial found the vaccine to be safe, with only six adverse events (AEs) reported by 39 participants, all grade 1, and effective, with all but three participants of 38 developing serum IgG binding titers to the SARS-CoV-2 S protein (1512).
In the phase II trial of 401 adults at high risk of exposure to SARS-CoV-2 similarly supported the safety and efficacy of INO-4800.
Only one treatment-related AE was observed and the vaccine was found to be associated with a significant increase in neutralizing activity (1513).
Results of phase III trials are not yet available (1514–1517).
Trials of ZyCoV-D have progressed further.
This vaccine uses a plasmid to deliver the expression-competent Spike protein and IgE signal peptides to the vaccinee (1518).
During the phase I trial, vaccination with a needle versus a needle-free injection system was evaluated, and the vaccine can now be administered without a needle (1506, 1507).
A phase III trial enrolling over 27,000 patients found no difference in AEs between the placebo and treatment groups and estimated the efficacy of ZyCoV-D to be 66.6% (1519).
It was authorized for people ages 12 and older (1508)
The highly portable design offers advantages over traditional vaccines (1518), especially as the emergence of variants continues to challenge the effectiveness of vaccines.
As of August 2022, ZyCoV-D has only been approved in India (1520) and is not tracked by Our World in Data (1116).
8.8.1.4 Real-World Safety and Effectiveness
In terms of the ability of plasmid-vectored vaccines to neutralize VOC, varying information is available.
The situation for ZyCoV-D is somewhat different, as their phase III trial occurred during the Delta wave in India (1519).
At present, no major press releases have addressed the vaccine’s ability to neutralize Omicron and related VOC, but reporting suggests that the manufacturers were optimistic about the vaccine in light of the Omicron variant as of late 2021 (1521).
As for INO-4800, studies have examined whether the induced immune response can neutralize existing VOC.
They assessed neutralization of several VOC relative to the index strain and found no difference in neutralization between the index strain and the Gamma VOC (P.1) (1522).
However, neutralization of the Alpha and Beta VOC was significantly lower (approximately two and seven times, respectively) (1522).
These findings are in line with the shifts in effectiveness reported for other vaccines (5).
In addition to loss of neutralizing activity due to viral evolution, studies have also evaluated the decline in neutralizing antibodies (nAbs) induced by INO-4800 over time.
Levels of nAbs remained statistically significant relative to the pre-vaccination baseline for six months (1523).
Administration of a booster dose induced a significant increase of titers relative to their pre-booster levels (1523).
Given the timing of this trial (enrollment between April and July 2020), it is unlikely that participants were exposed to VOC associated with decreased efficacy.
In light of the emergence of VOC against which many vaccines show lower effectiveness, Inovio Pharmaceuticals began to develop a new vaccine with the goal of improving robustness against known and future VOC (1524).
Known as INO-4802, this vaccine was designed to express a pan-Spike immunogen (1525).
Booster studies in rodents (1526) and non-human primates (1525) suggest that it may be more effective than INO-4800 in providing immunity to VOC such as Delta and Omicron when administered as part of a heterologous boost regimen, although boosting with INO-4800 was also very effective in increasing immunity in rhesus macaques (1525).
Therefore, boosting is likely to be an important strategy for this vaccine, especially as the virus continues to evolve.
8.8.2 Viral-Vectored DNA Vaccines
Plasmids are not the only vector that can be used to deliver sequences associated with viral antigens.
Genetic material from the target virus can also be delivered using a second virus as a vector.
Viral vectors have emerged as a safe and efficient method to furnish the nucleotide sequences of an antigen to the immune system (1527).
The genetic content of the vector virus is often altered to prevent it from replicating, but replication-competent viruses can also be used under certain circumstances (1528).
Once the plasmid or viral vector brings the DNA sequence to an APC, the host machinery can be used to construct antigen(s) from the transported genetic material, and the host can then synthesize antibodies in response (1479).
One of the early viral vectors explored was adenovirus, with serotype 5 (Ad5) being particularly effective (1479).
This technology rose in popularity during the 2000s due to its being more immunogenic in humans and non-human primates than plasmid-vectored DNA vaccines (1479).
In the 2000s, interest also arose in utilizing simian adenoviruses as vectors because of the reduced risk that human vaccine recipients would have prior exposure resulting in adaptive immunity (1479, 1529), and chimpanzee adenoviruses were explored as a potential vector in the development of a vaccine against MERS-CoV (1530).
Today, various viral-vector platforms including poxviruses (1531, 1532), adenoviruses (1533), and vesicular stomatitis viruses (1534, 1535) are being developed,
Viral-vector vaccines are able to induce both an antibody and cellular response; however, the response is limited due to the immunogenicity of the viral vector used (1533, 1536).
An important consideration in identifying potential vectors is the immune response to the vector.
Both the innate and adaptive immune responses can potentially respond to the vector, limiting the ability of the vaccine to transfer information to the immune system (1537).
Different vectors are associated with different levels of reactogenicity; for example, adenoviruses elicit a much stronger innate immune response than replication deficient adeno-associated viruses derived from parvoviruses (1537).
Additionally, using a virus circulating widely in human populations as a vector presents additional challenges because vaccine recipients may already have developed an immune response to the vector (1538).
Furthermore, repeated exposure to adenoviruses via viral-vectored DNA vaccines may increase reactivity to these vectors over time, presenting a challenge that will need to be considered in long-term development of these vaccines (1539, 1540).
8.8.2.1 Prior Applications
There are several viral vector vaccines that are available for veterinary use (1479, 1541), but prior to the COVID-19 pandemic, only one viral vector vaccine was approved by the United States’ Food and Drug Administration (FDA) for use in humans.
This vaccine is vectored with a recombinant vesicular stomatitis virus and targeted against the Ebola virus (1542).
Additionally, several phase I and phase II clinical trials for other vaccines are ongoing (1527), and the technology is currently being explored for its potential against numerous infectious diseases including malaria (1543, 1544), Ebola (1545–1547), and HIV (1548, 1549).
The threat of MERS and SARS initiated interest in the application of viral vector vaccines to human coronaviruses (1530), but efforts to apply this technology to these pathogens had not yet led to a successful vaccine candidate.
In the mid-to-late 2000s, adenoviral vectored vaccines against SARS were found to induce SARS-CoV-specific IgA in the lungs of mice (1550) but were later found to offer incomplete protection in ferret models (1551).
Gamaleya National Center of Epidemiology and Microbiology in Moscow sought to use an adenovirus platform for the development of vaccines for MERS-CoV and Ebola virus, although neither of the previous vaccines were internationally licensed (1552).
In 2017, results were published from an initial investigation of two vaccine candidates against MERS-CoV containing the MERS-CoV S gene vectored with chimpanzee adenovirus, Oxford University #1 (ChAdOx1), a replication-deficient chimpanzee adenovirus (1553).
This study reported that a candidate containing the complete S protein sequence induced a stronger neutralizing antibody response in mice than candidates vectored with modified vaccinia virus Ankara.
The candidate was pursued in additional research, and in the summer of 2020 results of two studies were published.
The first reported that a single dose of ChAdOx1 MERS induced an immune response and inhibited viral replication in macaques (1554).
The second reported promising results from a phase I trial that administered the vaccine to adults and measured safety, tolerability, and immune response (1555).
8.8.2.2 Application to COVID-19
Figure 11:Worldwide availability of vaccines developed using non-replicating viral vectors.
This figure reflects the number of vaccines using non-replicating viral vectors that were available in each country as of May 31, 2023.
These data are retrieved from Our World in Data (1116) and plotted using geopandas (1170).
See https://greenelab.github.io/covid19-review/ for the most recent version of this figure, which is updated daily.
Note that this figure draws from a different data source than Table 4 and does not necessarily include data for every vaccine developed within this category.
While not all of the above results were available at the time that vaccine development programs against SARS-CoV-2 began, at least three viral vector vaccines have also been developed against SARS-CoV-2 (Figure 11).
First, a collaboration between AstraZeneca and researchers at the University of Oxford successfully applied a viral vector approach to the development of a vaccine against SARS-CoV-2 using the replication-deficient ChAdOx1 vector modified to encode the S protein of SARS-CoV-2 (1556).
In a phase I trial, the immunogenic potential of vaccine candidate ChAdOx1 nCoV-19 was demonstrated through the immune challenge of two animal models, mice and rhesus macaques (1556).
In a phase I/II trial, patients receiving the ChAdOx1 nCoV-19 vaccine developed antibodies to the SARS-CoV-2 Spike protein that peaked by day 28, with these levels remaining stable until a second observation at day 56 (1557).
Second, a viral vector approach was applied by Russia’s Gamaleya Research Institute of Epidemiology and Microbiology to develop Sputnik V, a replication-deficient recombinant adenovirus (rAd) vaccine that combines two adenovirus vectors, rAd26-S and rAd5-S, that express the full-length SARS-CoV-2 Spike glycoprotein.
These vectors are intramuscularly administered individually using two separate vaccines in a prime-boost regimen.
The rAd26-S is administered first, followed by rAd5-S 21 days later.
Both vaccines deliver 1011 viral particles per dose.
This approach is designed to overcome any potential pre-existing immunity to adenovirus in the population (1558), as some individuals may possess immunity to Ad5 (1559).
Sputnik V is the only recombinant adenovirus vaccine to utilize two vectors.
Third, Janssen Pharmaceuticals, Inc., a subsidiary of Johnson & Johnson, developed a viral vector vaccine in collaboration with and funded by the United States’ “Operation Warp Speed” (1560, 1561).
The vaccine candidate JNJ-78436735, formerly known as Ad26.COV2-S, is a monovalent vaccine that is composed of a replication-deficient adenovirus serotype 26 (Ad26) vector expressing the stabilized prefusion S protein of SARS-CoV-2 (1485, 1562).
Unlike the other two viral vector vaccines available to date, JNJ-78436735 requires only a single dose, a characteristic that was expected to aid in global deployment (1563).
JNJ-78436735 was selected from among a number of initial candidate designs (1485) and tested in vivo in Syrian golden hamsters and Rhesus macaques to assess safety and immunogenicity (1485, 1563–1565).
The JNJ-78436735 candidate was selected for its favorable immunogenicity profile and ease of manufacturability (1485, 1563–1565) and was found to confer protection against SARS-CoV-2 in macaques even after six months (1566).
The one- versus two-dose regimen was then tested in volunteers through a phase I/IIa trial (1562, 1567).
A major difference between this vaccine and the other two in this category is that the S protein immunogen is stabilized in its prefusion conformation, while in the Sputnik V and AstraZeneca vaccines it is not.
As of May 31, 2023, data describing the distribution of 5 viral-vectored vaccines in 203 countries are available (Figure 11).
ChAdOx1 nCoV-19 was first approved for emergency use on December 30, 2020 in the U.K. (1568).
Sputnik V was available soon after, and early as January 2021, Sputnik V had been administered to 1.5 million Russians (1569) and began distributing doses to other countries within Europe such as Belarus, Bosnia-Herzegovina, Hungary, San Marino, Serbia, and Slovakia (1570–1572).
8.8.2.3 Trial Estimates of Safety and Efficacy
The first DNA viral-vectored vaccine for which efficacy estimates became available was AstraZeneca’s ChAdOx1 nCoV-19.
In December 2020, preliminary results of the phase III trial were released detailing randomized control trials conducted in the United Kingdom (U.K.), Brazil, and South Africa between April and November 2020 (1469).
These trials compared ChAdOx1 nCoV-19 to a control, but the design of each study varied; pooling data across studies indicated an overall efficacy of 70.4%.
For Sputnik V, the phase III trial indicated an overall vaccine efficacy of 91.6% for symptomatic COVID-19 (1573).
As for Janssen, the vaccine was well-tolerated, and across all regions studied, it was found to be 66.9% effective after 28 days for the prevention of moderate to severe COVID-19 and to be 81.7% effective for the prevention of laboratory-confirmed severe COVID-19 (1574).
There were no COVID-19-associated deaths in the vaccine group.
However, the emergence of the Beta variant in the South African trial population was associated with a slightly reduced efficacy (64% two weeks after receipt), and all of the COVID-19-associated deaths in the trial occurred in the South African placebo cohort (1574).
In February 2021, the FDA issued an EUA for the Janssen vaccine based on interim results from the phase III trial (1575, 1576).
Two of the three vaccines have faced a number of criticisms surrounding the implementation of their clinical trials.
In the race to develop vaccines against SARS-CoV-2, President Vladimir Putin of Russia announced the approval of the Sputnik V vaccines on August 11, 2020 in the absence of clinical evidence (1577).
A press release on November 11, 2020 indicated positive results from an interim analysis of the phase III Sputnik V trials, which reported 92% efficacy in 16,000 volunteers (1578).
However, this release came only two days after both Pfizer and BioNTech reported that their vaccines had an efficacy over 90%, which led to significant skepticism of the Russian findings for a myriad of reasons including the lack of a published protocol and the “reckless” approval of the vaccine in Russia months prior to the publication of the interim results of the phase III trial (1578, 1579).
Consequently, many international scientific agencies and public health bodies expressed concern that due diligence to the clinical trial process was subverted for the sake of expediency, leading many to question the safety and efficacy of Sputnik V (1577, 1580, 1581).
Despite regulatory, safety, and efficacy concerns, pre-orders for 1 billion doses of the Sputnik V were reported within days of the vaccine’s approval in Russia (1577).
Almost a month later, the phase I/II trial data was published (1582)
It wasn’t until February 2021, six months after its approval in Russia, that interim results of the phase III trial were released (1573).
This publication reported a VE of 91% and a low rate of serious AEs, although there were several serious AEs that were determined not to be associated with the vaccine by an independent data monitoring committee about which little other information was released (1583).
AstraZeneca’s clinical trial also faced criticism.
The trial was paused in September 2020 following a severe adverse event in one participant (1584).
It was restarted soon after (1585), but it seems that the recent pause was not mentioned to the FDA during a call the morning before the story broke (1586).
Additionally, individual sites within the trial employed somewhat different designs but were combined for analysis.
For example, in South Africa, the trial was double-blinded, whereas in the U.K. and Brazil it was single-blinded, and one of the two trials carried out in the U.K. evaluated two dosing regimens (low dose or standard dose, both followed by standard dose).
Some of the trials used a meningococcal conjugate vaccine (MenACWY) as a control, while others used saline.
Data was pooled across countries for analysis, a design decision that was approved by regulators but raised some questions when higher efficacy was reported in a subgroup of patients who received a low-dose followed by a standard dose.
This group came about because some participants in the U.K. were erroneously primed with a much lower dose, which turned out to have higher efficacy than the intended dose (1587).
Combining the data then led to confusion surrounding the VE, as VE varied widely among conditions (e.g., 62% VE in the standard dose group vs 90% in the group that received a low prime dose (1469)).
Subsequent research, however, suggests that reducing the prime dose may, in fact, elicit a superior immune response in the long-term despite a lower initial response (1588).
Therefore, this error may serendipitously improve efficacy of vaccine-vectored vaccines broadly.
8.8.2.4 Real-World Safety and Efficacy
Following the trials, additional concerns have been raised about some of these vaccines.
Within a few days to a few weeks following their first dose of the AstraZeneca vaccine, three women developed extensive venous sinus thrombosis (1589).
In March 2021, administration of the vaccine was paused in several European countries while a possible link to thrombotic events was investigated (1590), as these adverse events had not been observed in clinical trials, but the European Medicine Agency (EMA) soon determined that 25 events were not related to the vaccine (1591).
The following month, the United States paused administration of the Janssen vaccine for ten days due to 15 similar AEs (1592, 1593), but the EMA, U.S. Centers for Disease Control, and the FDA’s Advisory Committee on Immunization Practices again identified the events as being very rare and the benefits of the vaccine as likely to outweigh its risks (1594–1597).
In Denmark and Norway, population-based estimates suggested AstraZeneca’s vaccine increased incidence of venous thromboembolic events by 11 cases over baseline per 100,000 doses (1598).
Estimates of the incidence in other western countries have also been low (1599).
In the US, thromboembolic events following the Janssen vaccine have also been very rare (1595).
Subsequently, a potential mechanism was identified: the adenovirus vector binding to platelet factor 4 (1600, 1601).
Because this adverse event is so rare, the risk is likely still outweighed by the risks associated with contracting COVID-19 (1602), which is also associated with thrombotic events) (1593, 1603).
Similarly, concerns about Guillain-Barré syndrome arose in connection to the Janssen vaccine, but these events have similarly been determined to be very rare and the benefits to outweigh the risks (1597).
Given that vaccines from multiple platforms are now widely available, people at increased risk of a specific severe AE may have options to pursue vaccination with a platform that does not carry such risks.
For example, a woman in the U.S. with a history of thromboembolic concerns might feel more comfortable with an mRNA vaccine (described below), where such AEs have not been identified in association with COVID-19 vaccination.
However, within the U.S.A., no clear framework has been established for advising patients on whether a specific vaccine may be preferable for their individual concerns now that vaccines based on three different technologies are widely available (see (5) for information about Novavax, which is a protein subunit vaccine).
8.9 mRNA Vaccines
Building on DNA vaccine technology, RNA vaccines are an even more recent advancement for vaccine development.
Interest in messenger RNA (mRNA) vaccines emerged around 1990 following in vitro and animal model studies that demonstrated that mRNA could be transferred into cells (1604, 1605).
mRNA contains the minimum information needed to create a protein (1605).
RNA vaccines are therefore nucleic-acid based modalities that code for viral antigens against which the human body elicits a humoral and cellular immune response.
The strategy behind mRNA vaccines operates one level above the DNA: instead of directly furnishing the gene sequence associated with an antigen to the host, it provides the mRNA transcribed from the DNA sequence.
The mRNA is transcribed in vitro and delivered to cells via lipid nanoparticles (LNP) (1606).
It is recognized by ribosomes in vivo and then translated and modified into functional proteins (1127).
The resulting intracellular viral proteins are displayed on surface MHC proteins, provoking a strong CD8+ T cell response as well as a CD4+ T cell and B cell-associated antibody responses (1127).
mRNA is naturally not very stable and can degrade quickly in the extracellular environment or the cytoplasm.
The LNP covering protects the mRNA from enzymatic degradation outside of the cell (1607).
Codon optimization to prevent secondary structure formation and modifications of the poly-A tail as well as the 5’ untranslated region to promote ribosomal complex binding can increase mRNA expression in cells.
Furthermore, purifying out double-stranded RNA and immature RNA with fast performance liquid chromatography and high performance liquid chromatography technology will improve translation of the mRNA in the cell (1127, 1608).
There are three types of RNA vaccines: non-replicating, in vivo self-replicating, and in vitro dendritic cell non-replicating (1609).
Non-replicating mRNA vaccines consist of a simple open reading frame for the viral antigen flanked by the 5’ UTR and 3’ poly-A tail.
In vivo self-replicating vaccines encode a modified viral genome derived from single-stranded, positive sense RNA alphaviruses (1127, 1608).
The RNA genome encodes the viral antigen along with proteins of the genome replication machinery, including an RNA polymerase.
Structural proteins required for viral assembly are not included in the engineered genome (1127).
Self-replicating vaccines produce more viral antigens over a longer period of time, thereby evoking a more robust immune response (1609).
Finally, in vitro dendritic cell non-replicating RNA vaccines limit transfection to dendritic cells.
Dendritic cells are potent antigen-presenting immune cells that easily take up mRNA and present fragments of the translated peptide on their MHC proteins, which can then interact with T cell receptors.
Ultimately, primed T follicular helper cells can stimulate germinal center B cells that also present the viral antigen to produce antibodies against the virus (1610).
These cells are isolated from the patient, then grown and transfected ex vivo(1611).
They can then be reintroduced to the patient (1611).
In addition to the benefits of nucleic acid vaccines broadly, mRNA confers specific advantages compared to DNA vaccines and other platforms (1612).
Some of these advantages fall within the domain of safety.
Unlike DNA vaccines, mRNA technologies are naturally degradable and non-integrating, and they do not need to cross the nuclear membrane in addition to the plasma membrane for their effects to be seen (1127).
Additionally, the half life can be regulated by the contents of the 5’ and 3’ untranslated regions (1613).
In comparison to vaccines that use live attenuated viruses, mRNA vaccines are non-infectious and can be synthetically produced in an egg-free, cell-free environment, thereby reducing the risk of a detrimental immune response in the host (1614).
Furthermore, mRNA vaccines are easily, affordably, and rapidly scalable, despite the fact that it took time to reach the scale needed to manufacture vaccines at a scale sufficient for the global population (1612).
8.9.0.1 Prior Applications
Although mRNA vaccines have been developed for therapeutic and prophylactic purposes, none have previously been licensed or commercialized.
Challenges were caused by the instability of mRNA molecules, the design requirements of an efficient delivery system, and the potential for mRNA to elicit either a very strong immune response or to stimulate the immune system in secondary ways (1476, 1615).
As of the 2010s, mRNA was still considered a promising technology for future advances in vaccine development (1605), but prior to 2020, no mRNA vaccines had been approved for use in humans, despite significant advances in the development of this technology (1611).
This approach showed promise in animal models and preliminary clinical trials for several indications, including rabies, coronavirus, influenza, and cytomegalovirus (1616).
Preclinical data previously identified effective antibody generation against full-length purified influenza hemagglutinin stalk-encoding mRNA in mice, rabbits, and ferrets (1617).
Similar immunological responses for mRNA vaccines were observed in humans in phase I and II clinical trials operated by the pharmaceutical-development companies Curevac and Moderna for rabies, flu, and zika (1608).
Positively charged bilayer LNPs carrying the mRNA attract negatively charged cell membranes, endocytose into the cytoplasm (1607), and facilitate endosomal escape.
LNPs can be coated with modalities recognized and engulfed by specific cell types, and LNPs that are 150 nm or less effectively enter into lymphatic vessels (1607, 1618).
Therefore, while these technologies elegantly capitalize on decades of research in vaccine development as well as the tools of the genomic revolution, it was largely unknown prior to the SARS-CoV-2 pandemic whether this potential could be realized in a real-world vaccination effort.
8.9.0.2 Application to COVID-19
Table 5: mRNA vaccines approved in at least one country (1169) as of May 3, 2023.
As a note, this table includes licensing of existing mRNA technology, i.e., TAK-919 is used to describe Takeda’s manufacturing of Moderna’s formulation.
Vaccine
Company
GEMCOVAC-19
Gennova Biopharmaceuticals Limited
Spikevax
Moderna
Spikevax Bivalent Original/Omicron BA.1
Moderna
Spikevax Bivalent Original/Omicron BA.4/BA.5
Moderna
Comirnaty
Pfizer/BioNTech
Comirnaty Bivalent Original/Omicron BA.1
Pfizer/BioNTech
Comirnaty Bivalent Original/Omicron BA.4/BA.5
Pfizer/BioNTech
TAK-919 (Moderna formulation)
Takeda
AWcorna
Walvax
Given the potential for mRNA technology to be quickly adapted for a new pathogen, it was favored as a potential vaccine against COVID-19, and fortunately, the prior work in mRNA vaccine development paid off, with 9 mRNA vaccines available in at least one country as of May 3, 2023 (Table 5).
In the vaccines developed under this approach, the mRNA coding for a stabilized prefusion Spike protein, which is immunogenic (1619), is furnished to the immune system in order to train its response.
Two vaccine candidates in this category emerged with promising phase III results at the end of 2020.
Both require two doses approximately one month apart.
The first was Pfizer/BioNTech’s BNT162b2, which contains the full prefusion stabilized, membrane-anchored SARS-CoV-2 Spike protein in a vaccine formulation based on modified mRNA (modRNA) technology (1620, 1621).
The second mRNA vaccine, mRNA-1273 developed by ModernaTX, is comprised by a conventional LNP-encapsulated RNA encoding a full-length prefusion stabilized S protein for SARS-CoV-2 (1622).
The vaccine candidates developed against SARS-CoV-2 using mRNA vectors utilize similar principles and technologies, although there are slight differences in implementation among candidates such as the formulation of the platform and the specific components of the Spike protein encapsulated (e.g., the full Spike protein vs. the RBD alone) (1623).
As of May 31, 2023, 2 mRNA vaccines are available in 169 countries (Figure 12).
Figure 12:Worldwide availability of vaccines developed using mRNA.
This figure reflects the number of vaccines based on mRNA technology that were available in each country as of May 31, 2023.
These data are retrieved from Our World in Data (1116) and plotted using geopandas (1170).
See https://greenelab.github.io/covid19-review/ for the most recent version of this figure, which is updated daily.
Note that this figure draws from a different data source than Table 5 and does not necessarily include data for every vaccine developed within this category.
The rapid and simultaneous development of these vaccines was met with some controversy related to intellectual property (IP).
First, the National Institutes of Health (NIH) and Moderna became involved in a patent dispute, after researchers at the NIH argued they were unfairly excluded from some patents filed based on their IP after they generated the stabilized modRNA sequence used in the vaccine (1624).
Ultimately, in late 2021, Moderna backed down on the patent application (1625).
However, in August 2022, the company filed their own suit against Pfizer/BioNTech over IP related to the modRNA used in the latter’s COVID-19 vaccine (1625, 1626).
The outcome of this suit remains to be seen.
8.9.0.3 Trial Safety and Immunogenicity
The VEs revealed by the Pfizer/BioNTech and Moderna clinical trials exceeded expectations.
In a phase II/III multinational trial, the Pfizer/BioNTech’s BNT162b2 vaccine was associated with a 95% efficacy against laboratory-confirmed COVID-19 and with mild-to-moderate local and systemic effects but a low risk of serious AEs when the prime-boost doses were administered 21 days apart (516).
The ModernaTX mRNA-1273 vaccine was the second mRNA vaccine to release phase III results, despite being the first mRNA vaccine to enter phase I clinical trials and publish interim results of their phase III trial a few months later.
Their study reported a 94.5% vaccine efficacy in preventing symptomatic COVID-19 in adults who received the vaccine at 99 sites around the United States (1627).
Similar to BNT162b2, the mRNA-1273 vaccine was associated with mild-to-moderate AEs but with a low risk of serious AEs (1627).
In late 2020, both vaccines received approval from the FDA under an emergency use authorization (1628, 1629), and these vaccines have been widely distributed, primarily in North America and the European Union (1186).
As the first mRNA vaccines to make it to market, these two highly efficacious vaccines demonstrate the power of this emerging technology, which has previously attracted scientific interest because of its potential to be used to treat non-infectious as well as infectious diseases.
8.9.0.4 Real-World Safety and Effectiveness
As vaccines were rolled out, one study sought to monitor their effectiveness in a real-world setting.
Between December 2020 and April 2021, this prospective cohort study obtained weekly nasal swabs from 3,975 individuals at high risk of SARS-CoV-2 exposure (health care workers, frontline workers, etc.) within the United States (332).
Among these participants, 3,179 (80%) had received at least one dose of an mRNA vaccine, and of those, 2,686 (84%) were fully vaccinated, corresponding to 68% of trial participants overall.
For each vaccinated participant (defined here as having received at least dose 1 more than 7 days ago) whose sample tested positive for SARS-CoV-2, they categorized the viral lineage(s) present in the sample as well as in samples from 3-4 unvaccinated individuals matched by site and testing date.
Overall efficacy of mRNA vaccines was estimated at 91% with full vaccination, similar to the reports from the clinical trials.
The occurrence of fevers was also lower in individuals who were partially or fully vaccinated, and the duration of symptoms was approximately 6 days shorter.
Among the five cases in fully vaccinated and 11 cases in partially vaccinated participants, the rate of infection by VOC was much higher than in the unvaccinated population (30% versus 10%), suggesting that the vaccine was less effective against the VOC than the index strain.
The WHO continues to monitor the emergence of variants and their impact on vaccine efficacy (529).
In general, mRNA vaccines remain highly effective against severe illness and death, but the effectiveness against infection generally has declined.
A study monitoring infections in a Minnesota cohort from January to July 2021 estimated that the effectiveness of the Moderna vaccine fell to 86% and Pfizer to 76%, although protection against hospitalization remained at 91% and 85%, respectively (1630).
In July of that year, as the Delta variant became dominant in the U.S.A., these estimates all fell, to an effectiveness of 76% for Moderna and 42% for Pfizer and effectiveness against hospitalization of 81% and 75%, respectively (1630).
With the emergence of the Omicron VOC, vaccine effectiveness has likely further declined.
A study in a diverse cohort in Southern California, U.S.A. found the effectiveness of the Moderna vaccine in participants who had received only the primary course to be 44% (1631).
A study in South Africa compared case and hospitalization records from a 4-week period where Omicron was dominant to a 2-month period where Delta was dominant and found that the effectiveness against hospitalization during the Omicron wave was approximately 70% compared to 93% during the Delta wave (1632).
Similarly, a large study in England of 2.5 million individuals suggested that not only the variants circulating, but also the time since vaccination, played a large role in vaccine effectiveness (1633).
Shortly after the BNT162b2 primary course, effectiveness against the Omicron VOC was as high as 65.5%, but this declined to below 10% by six months after the second dose.
For mRNA-1273, the decline was from 75.1% to 14.9%.
Therefore, it is unsurprising that in spite of vaccination programs, infection rates and hospitalization rates climbed in early 2022 in many Western countries including the United States (1634, 1635), especially given that many places simultaneously began to loosen public health restrictions designed to reduce viral spread.
On the side of safety, the only major concern that has been raised is a possible link between mRNA vaccination and myocarditis, especially in young men (1597).
This concern began with case reports of a small number of cases of myocarditis following vaccination in several countries (1636, 1637).
Following these reports, the Israeli Ministry of Health began surveillance to monitor the occurrence of myocarditis (1638).
They identified 283 cases, almost exactly half of which occurred following vaccination with Pfizer’s BNT162b2.
Close analysis of these cases determined that the vaccine did have a significant effect on the incidence of myocarditis; however, the rate of myocarditis remained low overall (1638).
The identification of young men as a population at particular risk of this AE was supported, and the risk was found to be greater after the second dose than the first.
Both this study and a study evaluating data collected from US population-based surveillance identified an increased risk with additional doses (1639).
However, most findings suggest that this AE does not have long-term negative effects; a 2021 meta-analysis identified 69 cases, all of which resulted in full recovery (1640).
Although these events are very rare, as with the possible thromboembolic AEs associated with viral-vectored DNA vaccines, these findings suggest that it may be prudent to offer a framework for decision making for patients particularly concerned about specific AEs in settings where multiple vaccines are available.
8.10 Booster Doses
Due to waning effectiveness of vaccines over time, especially in light of viral evolution, boosters have emerged as an important strategy in retaining the benefits of vaccination over time.
Booster shots are now recommended in many places, and boosters that account for multiple variants and strains of SARS-CoV-2 are now available in some places (531).
For example, in the U.S.A., the FDA recently recommended bivalent booster doses designed to account for the Omicron VOC (1641–1643).
In this case, bivalent refers to the fact that doses deliver both the original formulation and an updated vaccine designed for the Omicron subvariants circulating in summer 2022.
The fact that the FDA did not require additional clinical trials from manufacturers for Omicron subvariants BA.4 and BA.5 specifically suggests that the rapid authorization of strain changes in response to emerging VOC may be increasingly attainable (1644).
Results suggest that this fourth dose offered at least a short-term increase in VE against Omicron subvariants and also provided additional protection against hospitalization (1645).
Homologous booster doses have been investigated for most vaccines.
For example, over 14,000 adults were administered a booster (second) dose of the Janssen Ad26.COV2.S vaccine (1646).
The booster dose was highly efficacious, with severe COVID-19 and hospitalization prevented almost completely in the vaccinated group.
A booster dose was also found to improve immune response for Sputnik V vaccinees (1647).
For the AstraZeneca vaccine, a different approach was taken.
In the interest of distributing first doses as widely as possible, in some places the time between the first and second doses was extended.
One study assessed the immunogenicity and reactogenicity associated with delaying the second dose in the prime-boost series until up to 45 weeks after the first, reporting that an extended inter-dose period was associated with increased antibody titers 28 days after the second dose (1648).
This analysis also revealed that a third dose provided an additional boost in neutralizing activity (1648).
Third and fourth doses have been introduced for at least some populations in many places in response to the Omicron variant.
An early study in Israeli healthcare workers showed that the additional immunization was safe and immunogenic with antibody titers restored to peak-third dose titers.
No severe illness was reported in the cohort studied (274 versus 426 age-matched controls), and vaccine efficacy against infection was reported at 30% for BNT162b2 and 11% for mRNA-1273 (1649).
Other studies reported that a third dose of BNT162b2 raised vaccine effectiveness to 67.2% for approximately the first month but that the effectiveness dropped to 45.7% (1633).
Reduced and even low efficacy against infection does not undermine the value of vaccination, considering the vaccines are intended to prevent severe disease, hospitalization, and death rather than infection generally.
However, these findings do suggest that boosters will likely be needed as the virus continues to evolve.
Many trials have also investigated heterologous boosting approaches.
In particular, the mRNA vaccines are a popular choice for booster doses regardless of primary series.
In general, such approaches have been found to confer favorable immunogenicity relative to homologous boosters (e.g., (1650–1656) and many other studies).
Due to remaining concerns about rare thromboembolic events, vaccinees who received AstraZeneca for their primary course are advised in some countries to seek a heterologous booster (1657), although such guidances are not supported by the evidence, which indicates that the first dose of AstraZeneca is most likely to be linked to these rare events (1658).
In general, heterologous boosting with mRNA vaccines elicits a strong immune response.
For patients who received BNT162b2 as a heterologous booster following a ChAdOx1 primary series, the vaccine effectiveness was estimated to be 62.4% initially, dropping to 39.6% after 10 weeks (1633).
For a heterologous mRNA-1273 booster, the effectiveness was estimated to be slightly higher (70.1% and 60.9% following ChAdOx1 and 73.9% to 64.4% following BNT162b2) (1633).
Therefore, subsequent booster doses may remain an ongoing component of strategies to combat SARS-CoV-2.
Although the vaccines developed based on the index strain remain highly effective at preventing severe illness and death, they serve much less utility at preventing illness broadly than they did early in the pandemic.
Therefore, many manufacturers are exploring potential reformulations based on VOC that have emerged since the beginning of the pandemic.
In June 2022, Moderna released data describing the effect of their bivalent mRNA booster, mRNA-1273.214, designed to protect against the Omicron variant (1659).
A 50 μg dose of mRNA-1273.214 was administered to 437 participants.
One month later, the neutralizing geometric mean titer ratio was assessed against several variants of SARS-CoV-2, including Omicron.
The immune response was higher against all variants assessed, including Omicron, than for boosting with the original formulation (mRNA-1273).
Another formulation, mRNA-1273.211, developed based on the Beta variant, has been associated with durable protection as long as six months after dosing.
The associated publications suggest that this novel formulation offers significant protection against Omicron and other VOC (1660, 1661).
In August 2022, Pfizer also announced successful development of a new formulation effective against Omicron (1662).
Modularity has been proposed as one of the advantages to developing DNA and mRNA vaccines.
This design would allow for faster adaptation to viral evolution.
However, in the arms race against SARS-CoV-2, the vaccines are still lagging behind the virus.
This disadvantage may change as regulators become more familiar with these vaccines and as a critical mass of data is accumulated.
Given the apparent need for boosters, interest has also emerged in whether updated formulations of SARS-CoV-2 vaccines can be administered along with annual flu vaccines to improve immunity to novel variants.
8.11 Conclusions
COVID-19 has seen the coming-of-age of vaccine technologies that have been in development since the late 20th century but had never before been authorized for use.
Vaccines that employ DNA and RNA eliminate all concerns about potential infection due to the vaccine components.
The vaccines described above demonstrate the potential for these technologies to facilitate a quick response to an emerging pathogen.
Additionally, their efficacy in trials far exceeded expectations, especially in the case of RNA vaccines.
These technologies hold significant potential to drive improvements in human health over the coming years.
Traditional vaccine technologies were built on the principle of using either a weakened version of the virus or a fragment of the virus.
COVID-19 has highlighted the fact that in recent years, the field has undergone a paradigm shift towards reverse vaccinology.
Reverse vaccinology emphasizes a discovery-driven approach to vaccine development based on knowledge of the viral genome (1663).
This strategy was explored during development of a DNA vaccine against the Zika virus (1664).
Though the disease was controlled before the vaccine became available (1423), the response demonstrated the potential for modular technologies to facilitate a response to emerging viral threats (1664).
The potential for such vaccines to benefit the field of oncology has encouraged vaccine developers to invest in next-generation approaches, which has spurred the diversification of vaccine development programs (1479, 1665).
As a result, during the COVID-19 pandemic, these modular technologies have taken center stage in controlling a viral threat for the first time.
The safety and efficacy of vaccines that use these new technologies has exceeded expectations.
While there were rare reports of severe AEs such as myocarditis (mRNA platforms) and thromboembolic events (viral-vectored DNA platforms), widespread availability of both types of vaccines would allow individuals to choose (particularly relevant in this case because myocarditis has primarily been reported in men and thromboembolic events primarily in women).
Estimates of efficacy have varied widely, but in all cases are high.
Estimates of the efficacy of DNA vaccine platforms have typically fallen either in the range of approximately 67% (ZyCoV-D and Janssen) or 90% (Sputnik V).
AstraZeneca’s trial produced estimates in both ranges, with the standard dosage producing an efficacy of 62% and the lower prime dose producing a VE of 90%.
The mRNA vaccine trials were somewhat higher, with VE estimated at approximately 95% for both the Moderna and Pfizer/BioNTech clinical trials.
However, in all cases, the efficacy against severe illness and death were very high.
Therefore, all of these vaccines are useful tools for combating COVID-19.
Furthermore, the fact that vaccine efficacy is not a static value has become particularly salient, as real-world effectiveness has changed with location and over time.
COVID-19 vaccines have been challenged by the emergence of VOC.
These VOC generally carry genetic mutations that code for an altered Spike protein (i.e., the antigen), so the antibodies resulting from immunization with vaccines developed from the index strain neutralize them less effectively (1666, 1667).
Despite some reports of varying and reduced effectiveness or efficacy of the mRNA vaccines against the Alpha (B.1.1.7), Beta (B.1.351), and Delta (B.1.617.2) variants versus the original SARS-CoV-2 strain or the D614G variant (1668–1670), the greatest concern to date has been the Omicron variant (B.1.1.529), which was first identified in November 2021 (1667, 1671).
As of March 2022, the Omicron variant accounted for 95% of all infections sequenced in the United States (122) and was linked to an increased risk of SARS-CoV-2 reinfection (1666) and further infection of those who have been vaccinated with the mRNA vaccines (1672).
One of the downsides of this leap in vaccine technologies, however, is that they have largely been developed by wealthy countries, including countries in the European Union, the United States, the U.K., and Russia.
As a result, they are also largely available to residents of wealthy countries, primarily in Europe and North America.
Although the VE of DNA vaccines tends to be lower than that of mRNA vaccines (1673), they still provide excellent protection against severe illness and are much easier to distribute due to less complex demands for storage.
Efforts such as COVAX that aim to expand access to vaccines developed by wealthy countries have not been as successful as hoped (1674).
Fortunately, vaccine development programs using more established technologies have been undertaken in many middle-income countries, and those vaccines have been more accessible globally (5).
Additionally, efforts to develop new formulations of DNA vaccines in lower- and middle-income countries are increasingly being undertaken (1675).
The modular nature of nucleic acid-based vaccine platforms has opened a new frontier in responding to emerging viral illnesses.
The RNA vaccines received an EUA in only a few months more than it took to identify the pathogen causing SARS in 2002.
Given the variety of options available for preventing severe illness and death, it is possible that certain vaccines may be preferable for certain demographics (e.g., young women might choose an mRNA vaccine to entirely mitigate the very low risk of blood clots (1597)).
However, this option is likely only available to people in high-income countries.
In lower-income countries, access to vaccines broadly is a more critical issue.
Different vaccines may confer advantages in different countries, and vaccine development in a variety of cultural contexts is therefore important (1676).
Without widespread access to vaccines on the global scale, SARS-CoV-2 will continue evolving, presenting a threat to all nations.
9 Appendix: Additional Information about Novel Vaccine Platforms for COVID-19
9.1 Plasmid-Vectored DNA Vaccines
9.1.1 INO-4800
The phase I trial for INO-4800 began enrolling participants in April 2020 in Philadelphia, PA at the Perelman School of Medicine and at the Center for Pharmaceutical Research in Kansas City, MO.
This trial examined two different doses administered in a two-dose regimen (1512).
Among the 39 participants, only six AEs were reported and all were grade 1 (1512).
Efficacy was evaluated based on blood samples collected pre- and post-vaccination, and all but three participants of 38 included in the analysis were found to have serum IgG binding titers to the spike protein after vaccination (1512).
Results from the phase II trial were released as a preprint in May 2021 and reported findings based on administering INO-4800 to 401 adult volunteers at high risk of exposure to SARS-CoV-2 (1513).
The phase II results supported that the vaccine was safe, with 1,446 treatment-related AEs observed across 281 participants, all but one of which were grade 1 or grade 2.
The single grade 3 event was joint stiffness (1513).
The rates of AEs in the placebo group were not reported.
To assess the immunogenicity of INO-4800, pre- and post-vaccination blood samples were collected and evaluated for a humoral immune response to the spike protein, and the treatment group was identified to show significantly greater neutralizing activity than the placebo group (1513).
The phase II/III trials are ongoing in several countries, including the United States, Mexico, India, and Colombia (1514–1517).
Therefore, vaccine efficacy data from a large study population is not yet available.
9.2 Viral-Vectored DNA Vaccines
9.2.1 ChAdOx1 nCoV-19 (AstraZeneca)
Prior analyses of viral vector vaccines against human coronaviruses (HCoVs) had indicated that this approach showed potential for inducing an immune response, but little information was available about the effect on real-world immunity.
In the first phase of development, a candidate ChAdOx1 nCoV-19 was evaluated through the immune challenge of two animal models, mice and rhesus macaques (1556).
Animals in the treatment condition were observed to develop neutralizing antibodies specific to SARS-CoV-2 (both macaques and mice) and to show reduced clinical scores when exposed to SARS-CoV-2 (macaques) (1556).
Next, a phase I/II trial was undertaken using a single-blind, randomized controlled design (1557).
ChAdOx1 nCoV-19 and a control, the meningococcal conjugate vaccine MenACWY, were administered intramuscularly to adults ages 18 to 55 at five sites within the United Kingdom (U.K.) at a 1:1 ratio (n=543 and n=534, respectively).
All but ten participants received a single dose; this small group received a booster 28 days after their first dose of ChAdOx1 nCoV-19.
Commonly reported local adverse reactions included mild-to-moderate pain and tenderness at the injection site over the course of seven days, while the most common systemic adverse reactions were fatigue and headache; some patients reported severe adverse systemic effects.
The study also reported that many common reactions could be reduced through the administration of paracetamol (acetaminophen), and paracetamol was not found to reduce immunogenicity.
Patients receiving the ChAdOx1 nCoV-19 vaccine developed antibodies to the SARS-CoV-2 spike protein that peaked by day 28, with these levels remaining stable until a second observation at day 56 except in the ten patients who received a booster dose at day 28, in whom they increased by day 56.
Analysis of serum indicated that participants developed antibodies to both S and the RBD, and that 100% of them achieved neutralizing titers by day 28.
By day 35, the neutralization titers of vaccinated patients was comparable to that observed with plasma from convalescents.
This initial study therefore suggested that the vaccine was likely to confer protection against SARS-CoV-2, although analysis of its efficacy in preventing COVID-19 was not reported.
The primary outcome assessed was symptomatic, laboratory-confirmed COVID-19.
There were 131 cases observed among the 11,636 participants eligible for the primary efficacy analysis, corresponding to an overall efficacy of 70.4% (30 out of 5,807 in the vaccine arm and 101 out of 5,829 in the control arm); the 95.8% CI was reported as 54.8 to 80.6.
However, a higher efficacy was reported in the subgroup of patients who received a low-dose followed by a standard dose (90.0%, 95% CI 67.4 to 97·0).
A total of ten cases of severe COVID-19 resulting in hospitalization were observed among trial participants, and all of these occurred in patients in the control arm of the study.
In line with the previously reported safety profiling for this vaccine, serious adverse events were reported to be comparable across the two arms of the study, with only three events identified as potentially associated with the vaccine itself.
Additional data about the efficacy of this vaccine became available in a preprint released on March 2, 2021 (1677).
This report provided data describing the efficacy of ChAdOx1 nCoV-19, along with Pfizer/BioNTech’s BNT162b2, in the U.K. between December 8, 2020 and February 19, 2021 and specifically sought to evaluate the efficacy of the vaccine in the presence of a potentially more contagious variant of concern, B.1.1.7.
All participants in this study were age 70 or older and the efficacy was estimated to increase from 60% at 28 days after vaccination to 73% at 35 days after vaccination, although the standard error also increased over this time.
Therefore, preliminary results suggest that in a number of samples, this vaccine confers a high level of protection against SARS-CoV-2.
9.2.2 Sputnik-V (Gam-COVID-Vac and Gam-COVID-Vac-Lyo)
The vaccine Gam-COVID-Vac, nicknamed Sputnik V in reference to the space race and “V for vaccine”, was developed by the Gamaleya National Center of Epidemiology and Microbiology in Moscow.
The development of Sputnik V was financed by the Russian Direct Investment Fund (RDIF) (1577, 1678).
The Sputnik V vaccines are available in both a lyophilized (Gam-COVID-Vac-Lyo) and frozen form (Gam-COVID-Vac), which are stored at 2-8°C and -18°C respectively (1582).
The lyophilized vaccine is convenient for distribution and storage, particularly to remote or disadvantaged areas (1679).
In the phase I/II trial study conducted between late June and early August 2020, 76 participants (18-60 years old) were split into two groups of 38 participants, which were non-randomized in two hospitals in Russia.
In phase I, 9 patients received rAd26 and 9 patients received rAd5-S to assess safety over 28 days.
In phase II, at least 5 days after the completion of phase I, 20 patients received a prime-boost vaccination of rAd26-S on day 0 and rAd5-S on day 2, which was administered intramuscularly.
The phase I/II trial reported that both vaccines were deemed safe and well tolerated.
The most common adverse events reported were mild, such as pain at the injection site (58%), hypothermia (50%), headaches (42%), fatigue (28%), and joint and muscle pain (24%).
Seroconversion was observed in all participants three weeks post the second vaccination (day 42), and all participants produced antibodies to the SARS-CoV-2 glycoprotein.
RBD-specific IgG levels were high in both the frozen and lyophilized versions of the vaccine (14,703 and 11,143 respectively), indicating a sufficient immune response to both.
Three weeks post the second vaccination, the virus-neutralizing geometric mean antibody titers were 49.25 and 45.95 from the frozen and lyophilized vaccines, respectively.
At 28 days, median cell proliferation of 1.3% CD4+ and 1.1% CD8+ were reported for the lyophilized vaccine and 2.5% CD4+ and 1.3% CD8+ for the vaccine stored frozen.
These results indicated that both forms of Sputnik V appeared to be safe and induced a humoral and cellular response in human subjects (1582), which may be robust enough to persist and not wane rapidly (1558).
In February 2021, the interim results of the phase III randomized, double-blind, placebo-controlled trial were published in The Lancet(1573).
The participants were randomly assigned to receive either a 0.5 mL/dose of vaccine or placebo, which was comprised of the vaccine buffer composition, that was delivered intramuscularly using the same prime-boost regimen as in the phase I/II trials.
From September 7 to November 24, 19,866 participants completed the trial.
Of the 14,964 participants who received the vaccine, 16 (0.1%) were confirmed to have COVID-19, whereas 62 of the 4,902 participants (1.3%) in the placebo group were confirmed to have COVID-19.
Of these participants, no moderate or severe cases of COVID-19 were reported in the vaccine group, juxtaposed with 20 in the placebo group.
However, only symptomatic individuals were confirmed for SARS-CoV-2 infection in this trial.
Therefore, asymptomatic infections were not detected, thus potentially inflating the efficacy estimate.
Overall, a vaccine efficacy of 91.6% (95% CI 85.6-95.2) was reported, where an efficacy of 91.8% was reported for those over 60 years old and 92.7% for those who were 51-60 years old.
Indeed, 14 days after the first dose, 87.6% efficacy was achieved and the immunity required to prevent disease occurred within 18 days of vaccination.
Based on these results, scientists are investigating the potential for a single dose regimen of the rAd26-S sputnik V vaccine (1680).
By the end of the trial, 7,485 participants reported adverse events, of which 94% were grade I.
Of the 68 participants who experienced serious adverse events during the trial, 45 from the vaccine group and 23 from the placebo groups, none were reported to be associated with the vaccination.
Likewise, 4 deaths occurred during the trial period that were not related to the vaccine (1573).
The interim findings of the phase III trial indicate that the Sputnik V vaccine regimen appears to be safe with 91.6% efficacy.
Gamaleya had intended to reach a total of 40,000 participants for the completion of their phase III trial.
However, the trial has stopped enrolling participants and the numbers have been cut to 31,000 as many individuals in the placebo group dropped out of the study to obtain the vaccine (1681).
Other trials involving Sputnik V are currently underway in Belarus, India, the United Arab Emirates, and Venezuela (1682).
Preliminary results of a trial of Argentinian healthcare workers in Buenos Aires who were vaccinated with the Sputnik V rAd26-R vector-based vaccine seems to support the short term safety of the first vaccination (1683).
Of the 707 vaccinated healthcare workers, 71.3% of the 96.6% of respondents reported at least one adverse event attributed to the vaccine.
Of these individuals, 68% experienced joint and muscle pain, 54% had injection site pain, 11% reported redness and swelling, 40% had a fever, and 5% reported diarrhea.
Only 5% of the vaccinated participants experienced serious adverse events that required medical attention, of which one was monitored as an inpatient.
Additionally, an independent assessment of Sputnik V in a phase II clinical trial in India found the vaccine to be effective, but the data is not yet publicly available (1684).
On December 21, 2020, Gamaleya, AstraZeneca, R-Pharm, and the Russian Direct Investment Fund agreed to assess the safety and immunogenicity of the combined use of components of the AstraZeneca and University of Oxford AZD1222 (ChAdOx1) vaccine and the rAd26-S component of the Sputnik V vaccine in clinical trials (1685).
This agreement hopes to establish scientific and business relations between the entities with an aim to co-develop a vaccine providing long-term immunization.
The trial, which will begin enrollment soon, will include 100 participants in a phase II open-label study and is hoped to be complete within 6 months.
Participants will first receive an intramuscular dose of AZD1222 on day 1, followed by a dose of rAd26 on day 29 and be monitored from day 1 for 180 days in total.
The primary outcomes measured will include incidence of serious adverse events post first dose until the end of the study.
Secondary outcome measures will include incidence of local and systemic adverse events 7 days post each dose, a time course of antibody responses for the Spike protein and the presence of anti-SARS-CoV-2 neutralizing antibodies (1686).
Overall, there is hesitancy surrounding the management of the Sputnik V vaccine approval process and concerns over whether the efficacy data may be inflated due to a lack of asymptomatic testing within the trial.
However, the interim results of the phase III study were promising and further trials are underway, which will likely shed light on the overall efficacy and safety of the Sputnik V vaccine regimen.
There may be some advantage to the Sputnik V approach including the favorable storage conditions afforded by choice between a frozen and lyophilized vaccine.
Furthermore, the producers of Gam-COVID-Vac state that they can produce the vaccine at a cost of less than $10 per dose or less than $20 per patient (1687).
9.3 Janssen’s JNJ-78436735
The Johnson and Johnson (J&J) vaccine developed by Janssen Pharmaceuticals, Inc., a subsidiary of J&J, was conducted in collaboration with and funded by “Operation Warp Speed” (1485, 1560–1562).
The vaccine was developed using Janssen’s AdVac® and PER.C6 platforms that were previously utilized to develop the European Commission-approved Ebola vaccine (Ad26 ZEBOV and MVN-BN-Filo) and their Zika, respiratory syncytial virus, and human immunodeficiency virus investigational vaccine candidates (1688).
The development of a single-dose vaccine was desired by J&J from the outset, with global deployment being a key priority (1563).
Using their AdVac® technology, the vaccine can remain stable for up to two years between -15 and -25℃ and at least three months at 2 to 8℃ (1688).
This allows the vaccine to be distributed easily without the requirement for very low temperature storage, unlike many of the other COVID-19 vaccine candidates.
J&J screened numerous potential vaccine candidates in vitro and in animal models using varying different designs of the S protein, heterologous signal peptides, and prefusion-stabilizing substitutions (1485).
A select few candidates were further investigated as a single dose regimen in Syrian golden hamsters, a single dose regimen in rhesus macaques, and a single- and two-dose regimen in both adult and aged rhesus macaques (1485, 1563–1565).
A SARS-CoV-2 challenge study in rhesus macaques showed that vaccine doses as low as 2 x 109 viral particles/mL was sufficient to induce strong protection in bronchoalveolar lavage but that doses higher than 1.125 x 1010 were required to close achieve close to complete protection in nasal swabs (1689).
Indeed, six months post-immunization, levels of S-binding and neutralizing antibodies in rhesus macaques indicated that the JNJ-78436735 vaccine conferred durable protection against SARS-CoV-2 (1566).
Following selection of the JNJ-78436735 vaccine, J&J began phase I/IIa trials.
The interim phase I/IIa data was placed on the medRxiv preprint server on September 25th, 2020 (1690) and was later published in the New England Journal of Medicine on January 13th, 2021 (1562).
The phase I/IIa multi-center, randomized, placebo-controlled trial enrolled 402 healthy participants between 18-55 years old and a further 403 healthy older participants ≥ 65 years old (1562).
Patients were administered either a placebo, a low dose (5 x 1010 viral particles per mL), or a high dose (1 X 1011 viral particles per mL) intramuscularly as part of either a single- or two-dose regimen.
All patients received injections 56 days apart, but participants in the single-dose condition received the placebo at the second appointment.
Those who received only one dose of either vaccine received a placebo dose at their second vaccination visit.
The primary endpoints of both the trial were safety and reactogenicity of each dose.
Fatigue, headache, myalgia, and pain at the injection site were the most frequent solicited adverse events reported by participants.
Although less common, particularly for those in the elderly cohort and those on the low dose regimen, the most frequent systemic adverse effect was fever.
Overall, immunization was well tolerated, particularly at the lower dose concentration.
In terms of reactogenicity, over 90% of those who received either the low or high dose demonstrated seroconversion in a neutralization assay using wild-type SARS-CoV-2, 29 days after immunization (1562).
Neutralizing geometric mean ratio of antibody titers (GMT) between 224-354 were detected regardless of age.
By day 57, 100% of the 18-55 year old participants had neutralizing GMT (288-488), which remained stable until day 71.
In the ≥ 65 years old cohort, the incidence of seroconversion for the low- and high-dose was 96% and 88% respectively by day 29.
GMTs for the low and high doses were slightly lower for participants ≥ 65 years old (196 and 127 respectively), potentially indicating slightly lower immunogenicity.
Seroconversion of the S antibodies was detected in 99% of individuals between 18-55 years old for the low and high doses (GMTs 528 and 695 respectively), with similar findings reported for the ≥ 65 years old.
Indeed, both dose concentrations also induced robust Th1 cytokine-producing S-specific CD4+ T cells and CD8+ T cell responses in both age groups.
The findings of the phase I/IIa study supported further investigation of a single immunization using the low dose vaccine.
Therefore, 25 patients were enrolled for a second randomized double-blind, placebo-controlled phase I clinical trial currently being conducted in Boston, Massachusetts for 2 years (1691).
Participants received either a single dose followed by a placebo, or a double dose of either a low dose (5 x 1010 viral particles/mL) or a high dose (1 x 1011 viral particles/mL) vaccine administered intramuscularly on day 1 or day 57.
Placebo-only recipients received a placebo dose on day 1 and 57.
Interim analyses conducted on day 71 indicated that binding and neutralizing antibodies developed 8 days after administration in 90% and 25% of vaccine recipients, respectively.
Binding and neutralizing antibodies were detected in 100% of vaccine recipients by day 57 after a single dose immunization.
Spike-specific antibodies were highly prevalent (GMT 2432 to 5729) as were neutralizing antibodies (GMT 242 to 449) in the vaccinated groups.
Indeed, CD4+ and CD8+ T-cell responses were also induced, which may provide additional protection, particularly if antibodies wane or poorly respond to infection (1692).
On September 23rd, 2020, J&J launched its phase III trial ENSEMBLE and released the study protocol to the public (1688, 1693).
The trial intended to enroll 60,000 volunteers to assess the safety and efficacy of the single vaccine dose versus placebo with primary endpoints of 14 and 28 days post-immunization (1688).
The trial was conducted in Argentina, Brazil, Chile, Colombia, Mexico, Peru, South Africa, and the U.S.
The trial was paused briefly in October 2020 to investigate a “serious medical event”, but resumed shortly after (1694).
An interim analysis was reported via press release on January 29th, 2021 (1575, 1576).
The interim data included 43,783 participants who accrued 468 symptomatic cases of COVID-19.
It was reported that the JNJ-78436735 vaccine was 66% effective across all regions studied for the prevention of moderate to severe COVID-19 28 days post-vaccination in those aged 18 years and older.
Notably, JNJ-78436735 was 85% effective for the prevention of laboratory-confirmed severe COVID-19 and 100% protection against COVID-19-related hospitalization and death 28 days post-vaccination across all study sites.
Efficacy of the vaccine against severe COVID-19 increased over time, and there were no cases of COVID-19 reported in immunized participants after day 49.
The trial also determined that the vaccine candidate has a favorable safety profile as determined by an independent Data and Safety Monitoring Board.
The vaccine was well tolerated, consistent with previous vaccines produced using the AdVac® platform.
Fever occurred in 9% of vaccine recipients, with grade 3 fever occurring in only 0.2% of recipients.
Serious adverse events were reportedly higher in the placebo group than the vaccine group, and no anaphylaxis was reported (1576).
At the time the phase III trial was being conducted, several concerning variants, including B.1.1.7 (482) and B.1.351 (231), were spreading across the globe.
In particular, B.1.351 was first identified in South Africa, which was one of the JNJ-78436735 vaccine trial sites.
Therefore, the J&J investigators also analyzed the efficacy of the JNJ-78436735 vaccine associated with their various trial sites to determine any potential risk of reduced efficacy as a result of the novel variants.
It was determined that JNJ-78436735 was 72% effective in the U.S., 66% effective in Latin America, and 57% effective in South Africa 28 days post-vaccination.
These findings underpin the importance of monitoring for the emergence of novel SARS-CoV-2 variants and determining their effects on vaccine efficacy.
Looking forward, Janssen are also running a phase III randomized, double-blind, placebo-controlled clinical trial, Ensemble 2, which aims to assess the efficacy, safety, and immunogenicity of a two-dose regimen of JNJ-78436735 administered 57 days apart.
This trial will enroll 30,000 participants ≥ 18 years old from Belgium, Colombia, France, Germany, Philippines, South Africa, Spain, U.K., and the U.S. (1695).
This trial will also include participants with and without comorbidities associated with an increased risk of COVID-19.
9.4 RNA Vaccines
RNA vaccines are nucleic-acid based modalities that code for viral antigens against which the human body elicits a humoral and cellular immune response.
The resulting intracellular viral proteins are displayed on surface MHC proteins, provoking a strong CD8+ T cell response as well as a CD4+ T cell and B cell-associated antibody responses (1127).
Given the potential for this technology to be quickly adapted for a new pathogen, it has held significant interest for the treatment of COVID-19.
The results of the interim analyses of two mRNA vaccine candidates became available at the end of 2020 and provided strong support for this emerging approach to vaccination.
Below we describe in detail the results available as of February 2021 for two such candidates, mRNA-1273 produced by ModernaTX and BNT162b2 produced by Pfizer, Inc. and BioNTech.
As of August 2022, the U.S. FDA has issues approvals or emergency use authorizations of versions of these vaccines for adults and for children 6 months and older (1696).
9.4.1 ModernaTX mRNA Vaccine
ModernaTX’s mRNA-1273 vaccine was the first COVID-19 vaccine to enter a phase I clinical trial in the United States.
An initial report described the results of enrolling forty-five participants who were administered intramuscular injections of mRNA-1273 in their deltoid muscle on day 1 and day 29, with the goal of following patients for the next twelve months (1119).
Healthy males and non-pregnant females aged 18-55 years were recruited for this study and divided into three groups receiving 25, 100, or 250 μg of mRNA-1273.
IgG ELISA assays on patient serology samples were used to examine the immunogenicity of the vaccine (1622).
Binding antibodies were observed at two weeks after the first dose at all concentrations.
At the time point one week after the second dose was administered on day 29, the pseudotyped lentivirus reporter single-round-of-infection neutralization assay, which was used to assess neutralizing activity, reached a median level similar to the median observed in convalescent plasma samples.
Participants reported mild and moderate systemic adverse events after the day 1 injection, and one severe local event was observed in each of the two highest dose levels.
The second injection led to severe systemic adverse events for three of the participants at the highest dose levels, with one participant in the group being evaluated at an urgent care center on the day after the second dose.
The reported localized adverse events from the second dose were similar to those from the first.
Several months later, a press release from ModernaTX described the results of the first interim analysis of the vaccine (1697).
On November 16, 2020, a report was released describing the initial results from phase III testing, corresponding to the first 95 cases of COVID-19 in the 30,000 enrolled participants (1697), with additional data released to the FDA on December 17, 2020 (1698).
These results were subsequently published in a peer-reviewed journal (The New England Journal of Medicine) on December 30, 2020 (1627).
The first group of 30,420 study participants was randomized to receive the vaccine or a placebo at a ratio of 1:1 (1627).
Administration occurred at 99 sites within the United States in two sessions, spaced 28 days apart (1627, 1699).
Patients reporting COVID-19 symptoms upon follow-up were tested for SARS-CoV-2 using a nasopharyngeal swab that was evaluated with RT-PCR (1699).
The initial preliminary analysis reported the results of the cases observed up until a cut-off date of November 11, 2020.
Of these first 95 cases reported, 90 occurred in participants receiving the placebo compared to 5 cases in the group receiving the vaccine (1697).
These results suggested the vaccine is 94.5% effective in preventing COVID-19.
Additionally, eleven severe cases of COVID-19 were observed, and all eleven occurred in participants receiving the placebo.
The publication reported the results through an extended cut-off date of November 21, 2020, corresponding to 196 cases (1627).
Of these, 11 occurred in the vaccine group and 185 in the placebo group, corresponding to an efficacy of 94.1%.
Once again, all of the severe cases of COVID-19 observed (n=30) occurred in the placebo group, including one death.
Thus, as more cases are reported, the efficacy of the vaccine has remained above 90%, and no cases of severe COVID-19 have yet been reported in participants receiving the vaccine.
These findings suggest the possibility that the vaccine might bolster immune defenses even for subjects who do still develop a SARS-CoV-2 infection.
The study was designed with an explicit goal of including individuals at high risk for COVID-19, including older adults, people with underlying health conditions, and people of color (1700).
The phase III trial population was comprised by approximately 25.3% adults over age 65 in the initial report and 24.8% in the publication (1699).
Among the cases reported by both interim analyses, 16-17% occurred in older adults (1627, 1697).
Additionally, approximately 10% of participants identified a Black or African-American background and 20% identified Hispanic or Latino ethnicity (1627, 1699).
Among the first 95 cases, 12.6% occurred in participants identifying a Hispanic or Latino background and 4% in participants reporting a Black or African-American background (1697); in the publication, they indicated only that 41 of the cases reported in the placebo group and 1 case in the treatment group occurred in “communities of color”, corresponding to 21.4% of all cases (1627).
While the sample size in both analyses is small relative to the study population of over 30,000, these results suggest that the vaccine is likely to be effective in people from a variety of backgrounds.
In-depth safety data was released by ModernaTX as part of their application for an EUA from the FDA and summarized in the associated publication (1627, 1699).
Because the detail provided in the report is greater than that provided in the publication, here we emphasize the results observed at the time of the first analysis.
Overall, a large percentage of participants reported adverse effects when solicited, and these reports were higher in the vaccine group than in the placebo group (94.5% versus 59.5%, respectively, at the time of the initial analysis) (1699).
Some of these events met the criteria for grade 3 (local or systemic) or grade 4 (systemic only) toxicity (1699), but most were grade 1 or grade 2 and lasted 2-3 days (1627).
The most common local adverse reaction was pain at the injection site, reported by 83.7% of participants receiving the first dose of the vaccine and 88.4% upon receiving the second dose, compared to 19.8% and 17.0%, respectively, of patients in the placebo condition (1699).
Fewer than 5% of vaccine recipients reported grade 3 pain at either administration.
Other frequent local reactions included erythema, swelling, and lymphadenopathy (1699).
For systemic adverse reactions, fatigue was the most common (1699).
Among participants receiving either dose of the vaccine, 68.5% reported fatigue compared to 36.1% participants receiving the placebo (1699).
The level of fatigue experienced was usually fairly mild, with only 9.6% and 1.3% of participants in the vaccine and placebo conditions, respectively, reporting grade 3 fatigue (1699), which corresponds to significant interference with daily activity (1701).
Based on the results of the report, an EUA was issued on December 18, 2020 to allow distribution of this vaccine in the United States (1629), and it was shortly followed by an Interim Order authorizing distribution of the vaccine in Canada (1702) and a conditional marketing authorization by the European Medicines Agency to facilitate distribution in the European Union (1703).
9.4.2 Pfizer/BioNTech BNT162b2
ModernaTX was, in fact, the second company to release news of a successful interim analysis of an mRNA vaccine and receive an EUA.
The first report came from Pfizer and BioNTech’s mRNA vaccine BNT162b2 on November 9, 2020 (1704), and a preliminary report was published in the New England Journal of Medicine one month later (516).
This vaccine candidate should not be confused with a similar candidate from Pfizer/BioNTech, BNT162b1, that delivered only the RBD of the spike protein (1705, 1706), which was not advanced to a phase III trial because of the improved reactogenicity/immunogenicity profile of BNT162b2 (517).
During the phase III trial of BNT162b2, 43,538 participants were enrolled 1:1 in the placebo and the vaccine candidate and received two 30-μg doses 21 days apart (516).
Of these enrolled participants, 21,720 received BNT162b2 and 21,728 received a placebo (516).
Recruitment occurred at 135 sites across six countries: Argentina, Brazil, Germany, South Africa, Turkey, and the United States.
An initial press release described the first 94 cases, which were consistent with 90% efficacy of the vaccine at 7 days following the second dose (1704).
The release of the full trial information covered a longer period and analyzed the first 170 cases occurring at least 7 days after the second dose, 8 of which occurred in patients who had received BNT162b2.
The press release characterized the study population as diverse, reporting that 42% of the participants worldwide came from non-white backgrounds, including 10% Black and 26% Hispanic or Latino (1707).
Within the United States, 10% and 13% of participants, respectively, identified themselves as having Black or Hispanic/Latino backgrounds (1707).
Additionally, 41% of participants worldwide were 56 years of age or older (1707), and they reported that the efficacy of the vaccine in adults over 65 was 94% (1708).
The primary efficacy analysis of the phase III study was concluded on November 18, 2020 (1708), and the final results indicted 94.6% efficacy of the vaccine (516).
The safety profile of the vaccine was also assessed (516).
A subset of patients were followed for reactogenicity using electronic diaries, with the data collected from these 8,183 participants comprising the solicited safety events analyzed.
Much like those who received the ModernaTX vaccine candidate, a large proportion of participants reported experiencing site injection pain within 7 days of vaccination.
While percentages are broken down by age group in the publication, these proportions correspond to approximately 78% and 73% of all participants after the first and second doses, respectively, overall.
Only a small percentage of these events (less than 1%) were rated as serious, with the rest being mild or moderate, and none reached grade 4.
Some participants also reported redness or swelling, and the publication indicates that in most cases, such events resolved within 1 to 2 days.
Participants also experienced systemic effects, including fever (in most cases lower than 38.9°C and more common after dose 2), fatigue (25-50% of participants depending on age group and dose), headache (25-50% of participants depending on age group and dose), chills, and muscle or joint pain; more rarely, patients could experience gastrointestinal effects such as vomiting or diarrhea.
As with the local events, these events were almost always grade 1 or 2.
While some events were reported by the placebo groups, these events were much rarer than in the treatment group even though compliance was similar.
Based on the efficacy and safety information released, the vaccine was approved in early December by the United Kingdom’s Medicines and Healthcare Products Regulatory Agency with administration outside of a clinical trial beginning on December 8, 2020 (1709, 1710).
As of December 11, 2020, the United States FDA approved this vaccine under an emergency use authorization (1628), and in August 2021, it received full approval for ages 16 and older (1711).
9.4.3 Neutralizing of VOC
Prior to studies examining the effectiveness of vaccines in real-world settings (summarized in (6), several studies reported reduced efficacy of the mRNA vaccines based on the measurement of antibody titers.
Plasma from individuals double-dosed with Pfizer/BioNTech’s BNT162b2 vaccine had up to a 16-fold reduction in neutralizing capacity against the Omicron variant (1712) and a reduced efficacy (70%) (1632).
Estimates for the mRNA vaccines range from a 2-fold to over a 20-fold drop in neutralisation titers (1713), hence the push for third and fourth doses of mRNA vaccines in many Western countries.
A third mRNA vaccine dose does increase antibody titers, but these levels also wane with time (1714).
Notably, immunocompromised individuals such as cancer patients seem to elicit a sufficient protective immune response against the Omicron variant when they have been boosted with a third dose of either mRNA vaccine, albeit a blunted response (1715).
While antibody titers do correlate with protection (1716–1720), they are not the only mechanisms of immune protection.
For example, T cell and non-neutralizing antibody responses may be unaffected or less affected by the new VOC, and they warrant further investigation.
9.5 Global Vaccine Status and Distribution
In North America, the majority of vaccines distributed until March 2021 have been produced by Pfizer-BioNTech and Moderna.
In Canada, the vaccine approval process is conducted by Health Canada, which uses a fast-tracked process whereby vaccine producers can submit data as it becomes available to allow for rapid review.
An approval may be granted following reviews of the available phase III clinical data.
This is followed by a period of pharmacovigilance in the population using their post-market surveillance system, which will monitor the long-term safety and efficacy of any vaccines (1721, 1722).
Health Canada has authorized the use of the Pfizer (December 9th, 2020), Moderna (December 23rd, 2020), Oxford-AstraZeneca (February 26th, 2021), and the Janssen (March 5th, 2021) vaccines, and the Novavax Inc vaccine is also under consideration (1723).
While Canada initially projected that by the end of September 2021 a vaccine would be available for all Canadian adults, they now predict that it may be possible earlier as more vaccines have been approved and become available (1724).
In the U.S., vaccines are required to have demonstrated safety and efficacy in phase III trials before manufacturers apply for an emergency use authorization (EUA) from the FDA.
If an EUA is granted, an additional evaluation of the safety and efficacy of the vaccines is conducted by the CDC’s Advisory Committee on Immunization Practices (ACIP) who also provide guidance on vaccine prioritization.
On December 1st, 2020, ACIP provided an interim phase 1a recommendation that healthcare workers and long-term care facility residents should be the first to be offered any vaccine approved (1725).
This was shortly followed by an EUA on December 11th, 2020 for the use of the Pfizer-BioNTech COVID vaccine (1726), which was distributed and administered to the first healthcare workers on December 14th, 2020 (1727).
Shortly thereafter, an EUA for the Moderna vaccine was issued on December 18th, 2020 (1728).
On December 20th, 2020, ACIP updated their initial recommendations to suggest that vaccinations should be offered to people aged 75 years old and older and to non-healthcare frontline workers in phase 1b (1729).
On the same date, it was recommended that phase 1c should include people aged 65-74 years old, individuals between the ages of 16-74 years old at high-risk due to health conditions, and essential workers ineligible in phase 1b (1729).
On the following day, December 21st, 2020, the first Moderna vaccines used outside of clinical trials were administered to American healthcare workers, which was the same day that President-elect Biden and Dr. Biden received their first doses of the Pfizer-BioNTech vaccine live on television to instill confidence in the approval and vaccination process (1730).
On February 27th, 2020, the FDA issued an EUA for the Janssen COVID-19 Vaccine (1731).
This was followed by an update on recommendations by ACIP for the use of the Janssen COVID-19 vaccine for those over 18 years old (1732).
The Janssen vaccine was first distributed to healthcare facilities on March 1st, 2021.
On March 12, 2021, the WHO added the Janssen vaccine to the list of safe and effective emergency tools for COVID-19 (1733).
While the CDC’s ACIP can provide recommendations, it is up to the public health authorities of each state, territory, and tribe to interpret the guidance and determine who will be vaccinated first (1734).
Prior to distribution of the Janssen vaccine, over 103 million doses of the Moderna and Pfizer-BioNTech vaccines were delivered across the U.S., with almost 79 million doses administered.
Of the total population, 15.6% have received at least one dose and 7.9% have received a second dose of either the Moderna (~38.3 million) or the Pfizer-BioNTech (~40.2 million) vaccines by February 28th, 2021 (1735).
President Biden’s administration has predicted that by the end of May 2021 there may be enough vaccine supply available for all adults in the U.S. (1736, 1737).
However, vaccine production, approval, and distribution was not straightforward in the U.S., as information was initially sparse and the rollout of vaccines was complicated by poor planning and leadership due to political activities prior to the change of administration in January 2021 (1738).
These political complications highlight the importance of the transparent vaccine approval process conducted by the FDA (1461).
Outside the U.S., the Moderna and Pfizer-BioNTech vaccines have been administered in 29 and 69 other countries, respectively, mainly in Europe and North America (1364).
The Janssen vaccine has so far only been administered in South Africa and the U.S. (1364, 1739), but it has also been approved in Bahrain, the European Union (E.U.), Iceland, Liechtenstein, and Norway (1186).
On March 11th, 2021, Johnson & Johnson received approval from the European Medicines Agency (EMA) for conditional marketing authorization of their vaccine (1740).
Notably, on March 2nd, 2021, rivals Johnson & Johnson and Merck announced that they entered an agreement to increase production of the Janssen vaccine to meet global demand (1741).
The U.K. was the first country to approve use of the Pfizer-BioNTech vaccine on December 2nd, 2020 (1742), and it was later approved by EMA on December 21st, 2020 (1743).
The U.K. was also the first to administer the Pfizer-BioNTech vaccine, making it the first COVID-19 vaccine supported by phase III data to be administered outside of clinical trials on December 8th, 2020.
The Oxford-AstraZeneca vaccine, was approved by the Medicines and Healthcare Products Regulatory Agency (MHRA) in the U.K. and by EMA in the E.U. on December 30th (2020) (1744) and January 29th (2021) (1602), respectively.
The Oxford-AstraZeneca vaccine was first administered in the UK on January 4th, 2021 (1745), and it is now being used in 53 countries in total, including Brazil, India, Pakistan, Mexico, and spanning most of Europe (1364).
The Moderna vaccine was authorized for use in the E.U. by EMA on January 6th, 2021 (1746) and in the U.K. by MHRA on January 8th, 2021 (1747).
As of March 5th, 2021, 22 million people in the U.K. had received at least one vaccine dose (1365).
While the Pfizer-BioNTech vaccine was the first to be distributed following phase III clinical trials, the first COVID-19 vaccine to be widely administered to people prior to the completion of phase III clinical trials was Sputnik V.
Sputnik V was administered to as many as 1.5 million Russians by early January (1569) due to the establishment of mass vaccination clinics in December 2020, prior to which only approximately 100,000 Russians had already been vaccinated (1748, 1749).
Doses of Sputnik V have also been distributed to other parts of Europe (1570–1572).
Hungary was the first E.U. member country to approve and distribute Sputnik V outside of Russia (1750), despite the EMA stating that they had neither approved nor received a request for approval of Sputnik V (1751).
Hungary is also in talks with China to procure the Sinopharm vaccines, which have been approved by Hungarian health authorities but also have not received approval by EMA in the E.U. (1750).
In Latin America, production facilities in both Brazil and Argentina will allow for increased production capacity of Sputnik V and doses have been distributed to Mexico, Argentina, Bolivia, Nicaragua, Paraguay, and Venezuela (1752).
Guinea was the first African nation to administer Sputnik V in December 2020, and the Central African Republic, Zimbabwe, and the Ivory Coast have all registered their interest in purchasing doses of the vaccine (1752).
In the Middle East, Iran has received its first doses of Sputnik V and the United Arab Emirates is conducting phase III trials (1752).
In Asia, while China’s vaccine candidates are favored, the Philippines, Nepal, and Uzbekistan have sought Sputnik V doses (1753).
In total, the RDIF claims to have received orders totalling 1.2 billion doses by over 50 countries worldwide (1753) and at least 18 countries are currently administering Sputnik V around the globe (1364).
Sputnik V has been an attractive vaccine for many countries due to its relatively low price, high efficacy, and its favorable storage conditions.
For some countries, Russia and China have also been more palatable politically than vaccine suppliers in the West (1752, 1754).
For others, the delays in the distribution of the other, more-favored candidates has been a motivating factor for pursuing the Sputnik V and Chinese alternatives (1571, 1754).
Additionally, Germany has stated that if Sputnik V were approved by EMA, it would be considered by the E.U. (1755).
Russia is developing other vaccine candidates and has approved a third vaccine, CoviVac, which is an inactivated vaccine produced by the Chumakov Centre in Moscow, despite the fact the clinical trials have yet to begin (1756).
10 Dietary Supplements and Nutraceuticals Under Investigation for COVID-19 Prevention and Treatment
10.1 Abstract
Coronavirus disease 2019 (COVID-19) has caused global disruption and a significant loss of life.
Existing treatments that can be repurposed as prophylactic and therapeutic agents could reduce the pandemic’s devastation.
Emerging evidence of potential applications in other therapeutic contexts has led to the investigation of dietary supplements and nutraceuticals for COVID-19.
Such products include vitamin C, vitamin D, omega 3 polyunsaturated fatty acids, probiotics, and zinc, all of which are currently under clinical investigation.
In this review, we critically appraise the evidence surrounding dietary supplements and nutraceuticals for the prophylaxis and treatment of COVID-19.
Overall, further study is required before evidence-based recommendations can be formulated, but nutritional status plays a significant role in patient outcomes, and these products could help alleviate deficiencies.
For example, evidence indicates that vitamin D deficiency may be associated with greater incidence of infection and severity of COVID-19, suggesting that vitamin D supplementation may hold prophylactic or therapeutic value.
A growing number of scientific organizations are now considering recommending vitamin D supplementation to those at high risk of COVID-19.
Because research in vitamin D and other nutraceuticals and supplements is preliminary, here we evaluate the extent to which these nutraceutical and dietary supplements hold potential in the COVID-19 crisis.
10.2 Importance
Sales of dietary supplements and nutraceuticals have increased during the pandemic due to their perceived “immune-boosting” effects.
However, little is known about the efficacy of these dietary supplements and nutraceuticals against the novel coronavirus (SARS-CoV-2) or the disease it causes, COVID-19.
This review provides a critical overview of the potential prophylactic and therapeutic value of various dietary supplements and nutraceuticals from the evidence available to date.
These include vitamin C, vitamin D, and zinc, which are often perceived by the public as treating respiratory infections or supporting immune health.
Consumers need to be aware of misinformation and false promises surrounding some supplements, which may be subject to limited regulation by authorities.
However, considerably more research is required to determine whether dietary supplements and nutraceuticals exhibit prophylactic and therapeutic value against SARS-CoV-2 infection and COVID-19.
This review provides perspective on which nutraceuticals and supplements are involved in biological processes that are relevant to recovery from or prevention of COVID-19.
10.3 Introduction
The year 2020 saw scientists and the medical community scrambling to repurpose or discover novel host-directed therapies against the coronavirus disease 2019 (COVID-19) pandemic caused by the spread of the novel Severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2).
This rapid effort led to the identification of some promising pharmaceutical therapies for hospitalized patients, such as remdesivir and dexamethasone.
Furthermore, most societies have adopted non-pharmacological preventative measures such as utilizing public health strategies that reduce the transmission of SARS-CoV-2.
However, during this time, many individuals sought additional protections via the consumption of various dietary supplements and nutraceuticals that they believed to confer beneficial effects.
While a patient’s nutritional status does seem to play a role in COVID-19 susceptibility and outcomes (1757–1761), the beginning of the pandemic saw sales of vitamins and other supplements soar despite a lack of any evidence supporting their use against COVID-19.
In the United States, for example, dietary supplement and nutraceutical sales have shown modest annual growth in recent years (approximately 5%, or a $345 million increase in 2019), but during the six-week period preceding April 5, 2020, they increased by 44% ($435 million) relative to the same period in 2019 (1762).
While growth subsequently leveled off, sales continued to boom, with a further 16% ($151 million) increase during the six weeks preceding May 17, 2020 relative to 2019 (1762).
In France, New Zealand, India, and China, similar trends in sales were reported (1763–1766).
The increase in sales was driven by a consumer perception that dietary supplements and nutraceuticals would protect consumers from infection and/or mitigate the impact of infection due to the various “immune-boosting” claims of these products (1767, 1768).
Due to the significant interest from the general public in dietary additives, whether and to what extent nutraceuticals or dietary supplements can provide any prophylactic or therapeutic benefit remains a topic of interest for the scientific community.
Nutraceuticals and dietary supplements are related but distinct non-pharmaceutical products.
Nutraceuticals are classified as supplements with health benefits beyond their basic nutritional value (1769, 1770).
The key difference between a dietary supplement and a nutraceutical is that nutraceuticals should not only supplement the diet, but also aid in the prophylaxis and/or treatment of a disorder or disease (1771).
However, dietary supplements and nutraceuticals, unlike pharmaceuticals, are not subject to the same regulatory protocols that protect consumers of medicines.
Indeed, nutraceuticals do not entirely fall under the responsibility of the Food and Drug Administration (FDA), but they are monitored as dietary supplements according to the Dietary Supplement, Health and Education Act 1994 (DSHEA) (1772) and the Food and Drug Administration Modernization Act 1997 (FDAMA) (1773).
Due to increases in sales of dietary supplements and nutraceuticals, in 1996 the FDA established the Office of Dietary Supplement Programs (ODSP) to increase surveillance.
Novel products or nutraceuticals must now submit a new dietary ingredient notification to the ODSP for review.
There are significant concerns that these legislations do not adequately protect the consumer as they ascribe responsibility to the manufacturers to ensure the safety of the product before manufacturing or marketing (1774).
Manufacturers are not required to register or even seek approval from the FDA to produce or sell food supplements or nutraceuticals.
Health or nutrient content claims for labeling purposes are approved based on an authoritative statement from the Academy of Sciences or relevant federal authorities once the FDA has been notified and on the basis that the information is known to be true and not deceptive (1774).
Therefore, there is often a gap between perceptions by the American public about a nutraceutical or dietary supplement and the actual clinical evidence surrounding its effects.
Despite differences in regulations, similar challenges exist outside of the United States.
In Europe, where the safety of supplements is monitored by the European Union (EU) under Directive 2002/46/EC (1775).
However, nutraceuticals are not directly mentioned.
Consequently, nutraceuticals can be generally described as either a medicinal product under Directive 2004/27/EC (1776) or as a ‘foodstuff’ under Directive 2002/46/EC of the European council.
In order to synchronize the various existing legislations, Regulation EC 1924/2006 on nutrition and health claims was put into effect to assure customers of safety and efficacy of products and to deliver understandable information to consumers.
However, specific legislation for nutraceuticals is still elusive.
Health claims are permitted on a product label only following compliance and authorization according to the European Food Safety Authority (EFSA) guidelines on nutrition and health claims (1777).
EFSA does not currently distinguish between food supplements and nutraceuticals for health claim applications of new products, as claim authorization is dependent on the availability of clinical data in order to substantiate efficacy (1778).
These guidelines seem to provide more protection to consumers than the FDA regulations but potentially at the cost of innovation in the sector (1779).
The situation becomes even more complicated when comparing regulations at a global level, as countries such as China and India have existing regulatory frameworks for traditional medicines and phytomedicines not commonly consumed in Western society (1780).
Currently, there is debate among scientists and regulatory authorities surrounding the development of a widespread regulatory framework to deal with the challenges of safety and health claim substantiation for nutraceuticals (1774, 1778), as these products do not necessarily follow the same rigorous clinical trial frameworks used to approve the use of pharmaceuticals.
Such regulatory disparities have been highlighted by the pandemic, as many individuals and companies have attempted to profit from the vulnerabilities of others by overstating claims in relation to the treatment of COVID-19 using supplements and nutraceuticals.
The FDA has written several letters to prevent companies marketing or selling products based on false hyperbolic promises about preventing SARS-CoV-2 infection or treating COVID-19 (1781–1783).
These letters came in response to efforts to market nutraceutical prophylactics against COVID-19, some of which charged the consumer as much as $23,000 (1784).
There have even been some incidents highlighted in the media because of their potentially life threatening consequences; for example, the use of oleandrin was touted as a potential “cure” by individuals close to the former President of the United States despite its high toxicity (1785).
Thus, heterogeneous and at times relaxed regulatory standards have permitted high-profile cases of the sale of nutraceuticals and dietary supplements that are purported to provide protection against COVID-19, despite a lack of research into these compounds.
Notwithstanding the issues of poor safety, efficacy, and regulatory oversight, some dietary supplements and nutraceuticals have exhibited therapeutic and prophylactic potential.
Some have been linked with reduced immunopathology, antiviral and anti-inflammatory activities, or even the prevention of acute respiratory distress syndrome (ARDS) (1767, 1786, 1787).
A host of potential candidates have been highlighted in the literature that target various aspects of the COVID-19 viral pathology, while others are thought to prime the host immune system.
These candidates include vitamins and minerals along with extracts and omega-3 polyunsaturated fatty acids (n-3 PUFA) (1788).
In vitro and in vivo studies suggest that nutraceuticals containing phycocyanobilin, N-acetylcysteine, glucosamine, selenium or phase 2 inductive nutraceuticals (e.g. ferulic acid, lipoic acid, or sulforaphane) can prevent or modulate RNA virus infections via amplification of the signaling activity of mitochondrial antiviral-signaling protein (MAVS) and activation of Toll-like receptor 7 (1789).
Phase 2 inductive molecules used in the production of nutraceuticals are known to activate nuclear factor erythroid 2–related factor 2 (Nrf2), which is a protein regulator of antioxidant enzymes that leads to the induction of several antioxidant enzymes, such as gamma-glutamylcysteine synthetase.
While promising, further animal and human studies are required to assess the therapeutic potential of these various nutrients and nutraceuticals against COVID-19.
For the purpose of this review, we have highlighted some of the main dietary supplements and nutraceuticals that are currently under investigation for their potential prophylactic and therapeutic applications.
These include n-3 PUFA, zinc, vitamins C and D, and probiotics.
10.4 n-3 PUFA
One category of supplements that has been explored for beneficial effects against various viral infections are the n-3 PUFAs (1788), commonly referred to as omega-3 fatty acids, which include eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA).
EPA and DHA intake can come from a diet high in fish or through dietary supplementation with fish oils or purified oils (1790).
Other, more sustainable sources of EPA and DHA include algae (1791, 1792), which can also be exploited for their rich abundance of other bioactive compounds such as angiotensin converting enzyme inhibitor peptides and antiviral agents including phycobiliproteins, sulfated polysaccharides, and calcium-spirulan (1793).
n-3 PUFAs have been investigated for many years for their therapeutic potential (1794).
Supplementation with fish oils is generally well tolerated (1794), and intake of n-3 PUFAs through dietary sources or supplementation is specifically encouraged for vulnerable groups such as pregnant and lactating women (1795, 1796).
As a result, these well-established compounds have drawn significant interest for their potential immune effects and therapeutic potential.
Particular interest has arisen in n-3 PUFAs as potential therapeutics against diseases associated with inflammation.
n-3 PUFAs have been found to modulate inflammation by influencing processes such as leukocyte chemotaxis, adhesion molecule expression, and the production of eicosanoids (1797, 1798).
This and other evidence indicates that n-3 PUFAs may have the capacity to modulate the adaptive immune response (1770, 1790, 1797); for example, they have been found to influence antigen presentation and the production of CD4(+) Th1 cells, among other relevant effects (1799).
Certainly, preliminary evidence from banked blood samples from 100 COVID-19 patients suggests that patients with a higher omega-3 index, a measure of the amount of EPA and DHA in red blood cells, had a lower risk of death due to COVID-19 (1800).
Interest has also arisen as to whether nutritional status related to n-3 PUFAs can also affect inflammation associated with severe disease, such as ARDS or sepsis (1801, 1802).
ARDS and sepsis hold particular concern in the treatment of severe COVID-19; an analysis of 82 deceased COVID-19 patients in Wuhan during January to February 2020 reported that respiratory failure (associated with ARDS) was the cause of death in 69.5% of cases, and sepsis or multi-organ failure accounted for 28.0% of deaths (742).
Research in ARDS prior to current pandemic suggests that n-3 PUFAs may hold some therapeutic potential.
One study randomized 16 consecutive ARDS patients to receive either a fish oil-enriched lipid emulsion or a control lipid emulsion (comprised of 100% long-chain triglycerides) under a double-blinded design (1803).
They reported a statistically significant reduction in leukotriene B4 levels in the group receiving the fish oil-enriched emulsion, suggesting that the fish oil supplementation may have reduced inflammation.
However, they also reported that most of their tests were not statistically significant, and therefore it seems that additional research using larger sample sizes is required.
A recent meta-analysis of 10 randomized controlled trials (RCTs) examining the effects of n-3 PUFAs on ARDS patients did not find evidence of any effect on mortality, although the effect on secondary outcomes could not be determined due to a low quality of evidence (1804).
However, another meta-analysis that examined 24 RCTs studying the effects of n-3 fatty acids on sepsis, including ARDS-induced sepsis, did find support for an effect on mortality when n-3 fatty acids were administered via enteral nutrition, although a paucity of high-quality evidence again limited conclusions (1805).
Therefore, despite theoretical support for an immunomodulatory effect of n-3 PUFAs in COVID-19, evidence from existing RCTs is insufficient to determine whether supplementation offers an advantage in a clinical setting that would be relevant to COVID-19.
Another potential mechanism that has led to interest in n-3 PUFAs as protective against viral infections including COVID-19 is its potential as a precursor molecule for the biosynthesis of endogenous specialized proresolving mediators (SPM), such as protectins and resolvins, that actively resolve inflammation and infection (1806).
SPM have exhibited beneficial effects against a variety of lung infections, including some caused by RNA viruses (1807, 1808).
Several mechanisms for SPM have been proposed, including preventing the release of pro-inflammatory cytokines and chemokines or increasing phagocytosis of cellular debris by macrophages (1809).
In influenza, SPM promote antiviral B lymphocytic activities (1810), and protectin D1 has been shown to increase survival from H1N1 viral infection in mice by affecting the viral replication machinery (1811).
It has thus been hypothesized that SPM could aid in the resolution of the cytokine storm and pulmonary inflammation associated with COVID-19 (1812, 1813).
Another theory is that some comorbidities, such as obesity, could lead to deficiencies of SPM, which could in turn be related to the occurrence of adverse outcomes for COVID-19 (1814).
However, not all studies are in agreement that n-3 PUFAs or their resulting SPM are effective against infections (1815).
At a minimum, the effectiveness of n-3 PUFAs against infections would be dependent on the dosage, timing, and the specific pathogens responsible (1816).
On another level, there is still the question of whether fish oils can raise the levels of SPM levels upon ingestion and in response to acute inflammation in humans (1817).
Currently, Karolinska University Hospital is running a trial that will measure the levels of SPM as a secondary outcome following intravenous supplementation of n-3 PUFAs in hospitalized COVID-19 patients to determine whether n-3 PUFAs provides therapeutic value (1818, 1819).
Therefore, while this mechanism provides theoretical support for a role for n-3 PUFAs against COVID-19, experimental support is still needed.
A third possible mechanism by which n-3 PUFAs could benefit COVID-19 patients arises from the fact that some COVID-19 patients, particularly those with comorbidities, are at a significant risk of thrombotic complications including arterial and venous thrombosis (105, 1820).
Therefore, the use of prophylactic and therapeutic anticoagulants and antithrombotic agents is under consideration (1821, 1822).
Considering that there is significant evidence that n-3 fatty acids and other fish oil-derived lipids possess antithrombotic properties and anti-inflammatory properties (1790, 1823, 1824), they may have therapeutic value against the prothrombotic complications of COVID-19.
In particular, concerns have been raised within the medical community about using investigational therapeutics on COVID-19 patients who are already on antiplatelet therapies due to pre-existing comorbidities because the introduction of such therapeutics could lead to issues with dosing and drug choice and/or negative drug-drug interactions (1821).
In such cases, dietary sources of n-3 fatty acids or other nutraceuticals with antiplatelet activities could hold particular value for reducing the risk of thrombotic complications in patients already receiving pharmaceutical antiplatelet therapies.
A new clinical trial (1825) is currently recruiting COVID-19 positive patients to investigate the anti-inflammatory activity of a recently developed, highly purified nutraceutical derivative of EPA known as icosapent ethyl (VascepaTM) (1826).
Other randomized controlled trials that are in the preparatory stages intend to investigate the administration of EPA and other bioactive compounds to COVID-19 positive patients in order to observe whether anti-inflammatory effects or disease state improvements occur (1827, 1828).
Finally, while there have been studies investigating the therapeutic value of n-3 fatty acids against ARDS in humans, there is still limited evidence of their effectiveness (1829).
It should be noted that the overall lack of human studies in this area means there is limited evidence as to whether these supplements could affect COVID-19 infection.
Consequently, the clinical trials that are underway and those that have been proposed will provide valuable insight into whether the anti-inflammatory potential of n-3 PUFAs and their derivatives can be beneficial to the treatment of COVID-19.
All the same, while the evidence is not present to draw conclusions about whether n-3 PUFAs will be useful in treating COVID-19, there is likely little harm associated with a diet rich in fish oils, and interest in n-3 PUFA supplementation by the general public is unlikely to have negative effects.
10.5 Zinc
Zinc is nutrient supplement that may exhibit some benefits against RNA viral infections.
Zinc is a trace metal obtained from dietary sources or supplementation and is important for the maintenance of immune cells involved in adaptive and innate immunity (1830).
Supplements can be administered orally as a tablet or as a lozenge and are available in many forms, such as zinc picolinate, zinc acetate, and zinc citrate.
Zinc is also available from dietary sources including meat, seafood, nuts, seeds, legumes, and dairy.
The role of zinc in immune function has been extensively reviewed (1830).
Zinc is an important signaling molecule, and zinc levels can alter host defense systems.
In inflammatory situations such as an infection, zinc can regulate leukocyte immune responses and modulate the nuclear factor kappa-light-chain-enhancer of activated B cells, thus altering cytokine production (1831, 1832).
In particular, zinc supplementation can increase natural killer cell levels, which are important cells for host defense against viral infections (1830, 1833).
As a result of these immune-related functions, zinc is also under consideration for possible benefits against COVID-19.
Adequate zinc intake has been associated with reduced incidence of infection (1834) and antiviral immunity (1835).
A randomized, double-blind, placebo-controlled trial that administered zinc supplementation to elderly subjects over the course of a year found that zinc supplementation decreased susceptibility to infection and that zinc deficiency was associated with increased susceptibility to infection (1834).
Clinical trial data supports the utility of zinc to diminish the duration and severity of symptoms associated with common colds when it is provided within 24 hours of the onset of symptoms (1836, 1837).
An observational study showed that COVID-19 patients had significantly lower zinc levels in comparison to healthy controls and that zinc-deficient COVID-19 patients (those with levels less than 80 μg/dl) tended to have more complications (70.4% vs 30.0%, p = 0.009) and potentially prolonged hospital stays (7.9 vs 5.7 days, p = 0.048) relative to patients who were not zinc deficient (1838).
In coronaviruses specifically, in vitro evidence has demonstrated that the combination of zinc (Zn2+) and zinc ionophores (pyrithione) can interrupt the replication mechanisms of SARS-CoV-GFP (a fluorescently tagged SARS-CoV-1) and a variety of other RNA viruses (1839, 1840).
Currently, there are over twenty clinical trials registered with the intention to use zinc in a preventative or therapeutic manner for COVID-19.
However, many of these trials proposed the use of zinc in conjunction with hydroxychloroquine and azithromycin (1841–1844), and it is not known how the lack of evidence supporting the use of hydroxychloroquine will affect investigation of zinc.
One retrospective observational study of New York University Langone hospitals in New York compared outcomes among hospitalized COVID-19 patients administered hydroxychloroquine and azithromycin with zinc sulfate (n = 411) versus hydroxychloroquine and azithromycin alone (n = 521).
Notably, zinc is the only treatment that was used in this trial that is still under consideration as a therapeutic agent due to the lack of efficacy and potential adverse events associated with hydroxychloroquine and azithromycin against COVID-19 (1845–1847).
While the addition of zinc sulfate did not affect the duration of hospitalization, the length of ICU stays or patient ventilation duration, univariate analyses indicated that zinc did increase the frequency of patients discharged and decreased the requirement for ventilation, referrals to the ICU, and mortality (1848).
However, a smaller retrospective study at Hoboken University Medical Center New Jersey failed to find an association between zinc supplementation and survival of hospitalized patients (1849).
Therefore, whether zinc contributes to COVID-19 recovery remains unclear.
Other trials are now investigating zinc in conjunction with other supplements such as vitamin C or n-3 PUFA (1828, 1850).
Though there is, overall, encouraging data for zinc supplementation against the common cold and viral infections, there is currently limited evidence to suggest zinc supplementation has any beneficial effects against the current novel COVID-19; thus, the clinical trials that are currently underway will provide vital information on the efficacious use of zinc in COVID-19 prevention and/or treatment.
However, given the limited risk and the potential association between zinc deficiency and illness, maintaining a healthy diet to ensure an adequate zinc status may be advisable for individuals seeking to reduce their likelihood of infection.
10.6 Vitamin C
Vitamins B, C, D, and E have also been suggested as potential nutrient supplement interventions for COVID-19 (1788, 1851).
In particular vitamin C has been proposed as a potential therapeutic agent against COVID-19 due to its long history of use against the common cold and other respiratory infections (1852, 1853).
Vitamin C can be obtained via dietary sources such as fruits and vegetables or via supplementation.
Vitamin C plays a significant role in promoting immune function due to its effects on various immune cells.
It affects inflammation by modulating cytokine production, decreasing histamine levels, enhancing the differentiation and proliferation of T- and B-lymphocytes, increasing antibody levels, and protecting against the negative effects of reactive oxygen species, among other effects related to COVID-19 pathology (1854–1856).
Vitamin C is utilized by the body during viral infections, as evinced by lower concentrations in leukocytes and lower concentrations of urinary vitamin C.
Post-infection, these levels return to baseline ranges (1857–1861).
It has been shown that as little as 0.1 g/d of vitamin C can maintain normal plasma levels of vitamin C in healthy individuals, but higher doses of at least 1-3 g/d are required for critically ill patients in ICUs (1862).
Indeed, vitamin C deficiency appears to be common among COVID-19 patients (1863, 1864).
COVID-19 is also associated with the formation of microthrombi and coagulopathy (107) that contribute to its characteristic lung pathology (1865), but these symptoms can be ameliorated by early infusions of vitamin C to inhibit endothelial surface P-selectin expression and platelet-endothelial adhesion (1866).
Intravenous vitamin C also reduced D-dimer levels in a case study of 17 COVID-19 patients (1867).
D-dimer levels are an important indicator of thrombus formation and breakdown and are notably elevated in COVID-19 patients (103, 104).
There is therefore preliminary evidence suggesting that vitamin C status and vitamin C administration may be relevant to COVID-19 outcomes.
Larger-scale studies of vitamin C, however, have provided mixed results.
A recent meta-analysis found consistent support for regular vitamin C supplementation reducing the duration of the common cold, but that supplementation with vitamin C (> 200 mg) failed to reduce the incidence of colds (1868).
Individual studies have found Vitamin C to reduce the susceptibility of patients to lower respiratory tract infections, such as pneumonia (1869).
Another meta-analysis demonstrated that in twelve trials, vitamin C supplementation reduced the length of stay of patients in intensive care units (ICUs) by 7.8% (95% CI: 4.2% to 11.2%; p = 0.00003).
Furthermore, high doses (1-3 g/day) significantly reduced the length of an ICU stay by 8.6% in six trials (p = 0.003).
Vitamin C also shortened the duration of mechanical ventilation by 18.2% in three trials in which patients required intervention for over 24 hours (95% CI 7.7% to 27%; p = 0.001) (1862).
Despite these findings, an RCT of 167 patients known as CITRUS ALI failed to show a benefit of a 96-hour infusion of vitamin C to treat ARDS (1870).
Clinical trials specifically investigating vitamin C in the context of COVID-19 have now begun, as highlighted by Carr et al. (1853).
These trials intend to investigate the use of intravenous vitamin C in hospitalized COVID-19 patients.
The first trial to report initial results took place in Wuhan, China (1871).
These initial results indicated that the administration of 12 g/12 hr of intravenous vitamin C for 7 days in 56 critically ill COVID-19 patients resulted in a promising reduction of 28-day mortality (p = 0.06) in univariate survival analysis (1872).
Indeed, the same study reported a significant decrease in IL-6 levels by day 7 of vitamin C infusion (p = 0.04) (1873).
Additional studies that are being conducted in Canada, China, Iran, and the USA will provide additional insight into whether vitamin C supplementation affects COVID-19 outcomes on a larger scale.
Even though evidence supporting the use of vitamin C is beginning to emerge, we will not know how effective vitamin C is as a therapeutic for quite some time.
Currently (as of January 2021) over fifteen trials are registered with clinicaltrials.gov that are either recruiting, active or are currently in preparation.
When completed, these trials will provide crucial evidence on the efficacy of vitamin C as a therapeutic for COVID-19 infection.
However, the majority of supplementation studies investigate the intravenous infusion of vitamin C in severe patients.
Therefore, there is a lack of studies investigating the potential prophylactic administration of vitamin C via oral supplementation for healthy individuals or potentially asymptomatic SARS-CoV-2 positive patients.
Once again, vitamin C intake is part of a healthy diet and the vitamin likely presents minimal risk, but its potential prophylactic or therapeutic effects against COVID-19 are yet to be determined.
To maintain vitamin C status, it would be prudent for individuals to ensure that they consume the recommended dietary allowance of vitamin C to maintain a healthy immune system (1757).
The recommended dietary allowance according to the FDA is 75-90 mg/d, whereas EFSA recommends 110 mg/d (1874).
10.7 Vitamin D
Of all of the supplements currently under investigation, vitamin D has become a leading prophylactic and therapeutic candidate against SARS-CoV-2.
Vitamin D can modulate both the adaptive and innate immune system and is associated with various aspects of immune health and antiviral defense (1875–1879).
Vitamin D can be sourced through diet or supplementation, but it is mainly biosynthesized by the body on exposure to ultraviolet light (UVB) from sunlight.
Vitamin D deficiency is associated with an increased susceptibility to infection (1880).
In particular, vitamin D deficient patients are at risk of developing acute respiratory infections (1881) and ARDS (1881).
1,25-dihydroxyvitamin D3 is the active form of vitamin D that is involved in adaptive and innate responses; however, due to its low concentration and a short half life of a few hours, vitamin D levels are typically measured by the longer lasting and more abundant precursor 25-hydroxyvitamin D.
The vitamin D receptor is expressed in various immune cells, and vitamin D is an immunomodulator of antigen presenting cells, dendritic cells, macrophages, monocytes, and T- and B-lymphocytes (1880, 1882).
Due to its potential immunomodulating properties, vitamin D supplementation may be advantageous to maintain a healthy immune system.
Early in the pandemic it was postulated that an individual’s vitamin D status could significantly affect their risk of developing COVID-19 (1883).
This hypothesis was derived from the fact that the current pandemic emerged in Wuhan China during winter, when 25-hydroxyvitamin D concentrations are at their lowest due to a lack of sunlight, whereas in the Southern Hemisphere, where it was nearing the end of the summer and higher 25-hydroxyvitamin D concentrations would be higher, the number of cases was low.
This led researchers to question whether there was a seasonal component to the SARS-CoV-2 pandemic and whether vitamin D levels might play a role (1883–1886).
Though it is assumed that COVID-19 is seasonal, multiple other factors that can affect vitamin D levels should also be considered.
These factors include an individual’s nutritional status, their age, their occupation, skin pigmentation, potential comorbidities, and the variation of exposure to sunlight due to latitude amongst others.
Indeed, it has been estimated that each degree of latitude north of 28 degrees corresponded to a 4.4% increase of COVID-19 mortality, indirectly linking a persons vitamin D levels via exposure to UVB light to COVID-19 mortality (1884).
As the pandemic has evolved, additional research of varying quality has investigated some of the potential links identified early in the pandemic (1883) between vitamin D and COVID-19.
Indeed, studies are beginning to investigate whether there is any prophylactic and/or therapeutic relationship between vitamin D and COVID-19.
A study in Switzerland demonstrated that 27 SARS-CoV-2 positive patients exhibited 25-hydroxyvitamin D plasma concentrations that were significantly lower (11.1 ng/ml) than those of SARS-CoV-2 negative patients (24.6 ng/ml; p = 0.004), an association that held when stratifying patients greater than 70 years old (1887).
These findings seem to be supported by a Belgian observational study of 186 SARS-CoV-2 positive patients exhibiting symptoms of pneumonia, where 25-hydroxyvitamin D plasma concentrations were measured and CT scans of the lungs were obtained upon hospitalization (1888).
A significant difference in 25-hydroxyvitamin D levels was observed between the SARS-CoV-2 patients and 2,717 season-matched hospitalized controls.
It is not clear from the study which diseases caused the control subjects to be admitted at the time of their 25-hydroxyvitamin D measurement, which makes it difficult to assess the observations reported.
Both female and male patients possessed lower median 25-hydroxyvitamin D concentrations than the control group as a whole (18.6 ng/ml versus 21.5 ng/ml; p = 0.0016) and a higher rate of vitamin D deficiency (58.6% versus 42.5%).
However, when comparisons were stratified by sex, evidence of sexual dimorphism became apparent, as female patients had equivalent levels of 25-hydroxyvitamin D to females in the control group, whereas male patients were deficient in 25-hydroxyvitamin D relative to male controls (67% versus 49%; p = 0.0006).
Notably, vitamin D deficiency was progressively lower in males with advancing radiological disease stages (p = 0.001).
However, these studies are supported by several others that indicate that vitamin D status may be an independent risk factor for the severity of COVID-19 (1889–1892) and in COVID-19 patients relative to population-based controls (1893).
Indeed, serum concentrations of 25-hydroxyvitamin D above 30 ng/ml, which indicate vitamin D sufficiency, seems to be associated with a reduction in serum C-reactive protein, an inflammatory marker, along with increased lymphocyte levels, which suggests that vitamin D levels may modulate the immune response by reducing risk for cytokine storm in response to SARS-CoV-2 infection (1893).
A study in India determined that COVID-19 fatality was higher in patients with severe COVID-19 and low serum 25-hydroxyvitamin D (mean level 6.2 ng/ml; 97% vitamin D deficient) levels versus asymptomatic non-severe patients with higher levels of vitamin D (mean level 27.9 ng/ml; 33% vitamin D deficient) (1894).
In the same study, vitamin D deficiency was associated with higher levels of inflammatory markers including IL-6, ferritin, and tumor necrosis factor α.
Collectively, these studies add to a multitude of observational studies reporting potential associations between low levels of 25-hydroxyvitamin D and COVID-19 incidence and severity (1887, 1892, 1893, 1895–1901).
Despite the large number of studies establishing a link between vitamin D status and COVID-19 severity, an examination of data from the UK Biobank did not support this thesis (1902, 1903).
These analyses examined 25-hydroxyvitamin D concentrations alongside SARS-CoV-2 positivity and COVID-19 mortality in over 340,000 UK Biobank participants.
However, these studies have caused considerable debate that will likely be settled following further studies (1904, 1905).
Overall, while the evidence suggests that there is likely an association between low serum 25-hydroxyvitamin D and COVID-19 incidence, these studies must be interpreted with caution, as there is the potential for reverse causality, bias, and other confounding factors including that vitamin D deficiency is also associated with numerous pre-existing conditions and risk factors that can increase the risk for severe COVID-19 (1757, 1884, 1906, 1907).
While these studies inform us of the potential importance of vitamin D sufficiency and the risk of SARS-CoV-2 infection and severe COVID-19, they fail to conclusively determine whether vitamin D supplementation can therapeutically affect the clinical course of COVID-19.
In one study, 40 vitamin D deficient asymptomatic or mildly symptomatic participants patients were either randomized to receive 60,000 IU of cholecalciferol daily for at least 7 days (n = 16) or a placebo (n = 24) with a target serum 25-hydroxyvitamin D level >50 ng/ml.
At day 7, 10 patients achieved >50 ng/ml, followed by another 2 by day 14.
By the end of the study, the treatment group had a greater proportion of vitamin D-deficient participants that tested negative for SARS-CoV-2 RNA, and they had a significantly lower fibrinogen levels, potentially indicating a beneficial effect (1908).
A pilot study in Spain determined that early administration of high dose calcifediol (~21,000 IU days 1-2 and ~11,000 IU days 3-7 of hospital admission) with hydroxychloroquine and azithromycin to 50 hospitalized COVID-19 patients significantly reduced ICU admissions and may have reduced disease severity versus hydroxychloroquine and azithromycin alone (1909).
Although this study received significant criticism from the National Institute for Health and Care Excellence (NICE) in the UK (1910), an independent follow-up statistical analysis supported the findings of the study with respect to the results of cholecalciferol treatment (1911).
Another trial of 986 patients hospitalized for COVID-19 in three UK hospitals administered cholecalciferol (≥ 280,000 IU in a time period of 7 weeks) to 151 patients and found an association with a reduced risk of COVID-19 mortality, regardless of baseline 25-hydroxyvitamin D levels (1912).
However, a double-blind, randomized, placebo-controlled trial of 240 hospitalized COVID-19 patients in São Paulo, Brazil administered a single 200,000 IU oral dose of vitamin D.
At the end of the study, there was a 24 ng/mL difference of 25-hydroxyvitamin D levels in the treatment group versus the placebo group (p = 0.001), and 87% of the treatment group were vitamin D sufficient versus ~11% in the placebo group.
Supplementation was well tolerated.
However, there was no reduction in the length of hospital stay or mortality, and no change to any other relevant secondary outcomes were reported (1913).
These early findings are thus still inconclusive with regards to the therapeutic value of vitamin D supplementation.
However, other trials are underway, including one trial that is investigating the utility of vitamin D as an immune-modulating agent by monitoring whether administration of vitamin D precipitates an improvement of health status in non-severe symptomatic COVID-19 patients and whether vitamin D prevents patient deterioration (1914).
Other trials are examining various factors including mortality, symptom recovery, severity of disease, rates of ventilation, inflammatory markers such as C-reactive protein and IL-6, blood cell counts, and the prophylactic capacity of vitamin D administration (1914–1917).
Concomitant administration of vitamin D with pharmaceuticals such as aspirin (1918) and bioactive molecules such as resveratrol (1919) is also under investigation.
The effectiveness of vitamin D supplementation against COVID-19 remains open for debate.
All the same, there is no doubt that vitamin D deficiency is a widespread issue and should be addressed not only because of its potential link to SARS-CoV-2 incidence (1920), but also due to its importance for overall health.
There is a possibility that safe exposure to sunlight could improve endogenous synthesis of vitamin D, potentially strengthening the immune system.
However, sun exposure is not sufficient on its own, particularly in the winter months.
Indeed, while the possible link between vitamin D status and COVID-19 is further investigated, preemptive supplementation of vitamin D and encouraging people to maintain a healthy diet for optimum vitamin D status is likely to raise serum levels of 25-hydroxyvitamin D while being unlikely to carry major health risks.
These principles seem to be the basis of a number of guidelines issued by some countries and scientific organizations that have advised supplementation of vitamin D during the pandemic.
The Académie Nationale de Médecine in France recommends rapid testing of 25-hydroxyvitamin D for people over 60 years old to identify those most at risk of vitamin D deficiency and advises them to obtain a bolus dose of 50,000 to 100,000 IU vitamin D to limit respiratory complications.
It has also recommended that those under 60 years old should take 800 to 1,000 IU daily if they receive a SARS-CoV-2 positive test (1921/).
In Slovenia, doctors have been advised to provide nursing home patients with vitamin D (1922).
Both Public Health England and Public Health Scotland have advised members of the Black, Asian, and minority ethnic communities to supplement for vitamin D in light of evidence that they may be at higher risk for vitamin D deficiency along with other COVID-19 risk factors, a trend that has also been observed in the United States (1923, 1924).
However, other UK scientific bodies including the NICE recommend that individuals supplement for vitamin D as per usual UK government advice but warn that people should not supplement for vitamin D solely to prevent COVID-19.
All the same, the NICE has provided guidelines for research to investigate the supplementation of vitamin D in the context of COVID-19 (1925).
Despite vitamin D deficiency being a widespread issue in the United States (1926), the National Institutes of Health have stated that there is “insufficient data to recommend either for or against the use of vitamin D for the prevention or treatment of COVID-19” (1927/).
These are just some examples of how public health guidance has responded to the emerging evidence regarding vitamin D and COVID-19.
Outside of official recommendations, there is also evidence that individuals may be paying increased attention to their vitamin D levels, as a survey of Polish consumers showed that 56% of respondents used vitamin D during the pandemic (1928).
However, some companies have used the emerging evidence surrounding vitamin D to sell products that claim to prevent and treat COVID-19, which in one incident required a federal court to intervene and issue an injunction barring the sale of vitamin-D-related products due to the lack of clinical data supporting these claims (1929).
It is clear that further studies and clinical trials are required to conclusively determine the prophylactic and therapeutic potential of vitamin D supplementation against COVID-19.
Until such time that sufficient evidence emerges, individuals should follow their national guidelines surrounding vitamin D intake to achieve vitamin D sufficiency.
10.8 Probiotics
Probiotics are “live microorganisms that, when administered in adequate amounts, confer a health benefit on the host” (1930).
Some studies suggest that probiotics are beneficial against common viral infections, and there is modest evidence to suggest that they can modulate the immune response (1931, 1932).
As a result, it has been hypothesized that probiotics may have therapeutic value worthy of investigation against SARS-CoV-2 (1933).
Probiotics and next-generation probiotics, which are more akin to pharmacological-grade supplements, have been associated with multiple potential beneficial effects for allergies, digestive tract disorders, and even metabolic diseases through their anti-inflammatory and immunomodulatory effects (1934, 1935).
However, the mechanisms by which probiotics affect these various conditions would likely differ among strains, with the ultimate effect of the probiotic depending on the heterogeneous set of bacteria present (1935).
Some of the beneficial effects of probiotics include reducing inflammation by promoting the expression of anti-inflammatory mediators, inhibiting Toll-like receptors 2 and 4, competing directly with pathogens, synthesizing antimicrobial substances or other metabolites, improving intestinal barrier function, and/or favorably altering the gut microbiota and the brain-gut axis (1935–1937).
It is also thought that lactobacilli such as Lactobacillus paracasei, Lactobacillus plantarum and Lactobacillus rhamnosus have the capacity to bind to and inactivate some viruses via adsorptive and/or trapping mechanisms (1938).
Other probiotic lactobacilli and even non-viable bacterium-like particles have been shown to reduce both viral attachment to host cells and viral titers, along with reducing cytokine synthesis, enhancing the antiviral IFN-α response, and inducing various other antiviral mechanisms (1938–1946).
These antiviral and immunobiotic mechanisms and others have been reviewed in detail elsewhere (1787, 1933, 1947).
However, there is also a bi-directional relationship between the lungs and gut microbiota known as the gut-lung axis (1948), whereby gut microbial metabolites and endotoxins may affect the lungs via the circulatory system and the lung microbiota in return may affect the gut (1949).
Therefore, the gut-lung axis may play role in our future understanding of COVID-19 pathogenesis and become a target for probiotic treatments (1950).
Moreover, as microbial dysbiosis of the respiratory tract and gut may play a role in some viral infections, it has been suggested that SARS-CoV-2 may interact with our commensal microbiota [(1787); (1951); 10.3389/fmicb.2020.01840] and that the lung microbiome could play a role in developing immunity to viral infections (1952).
These postulations, if correct, could lead to the development of novel probiotic and prebiotic treatments.
However, significant research is required to confirm these associations and their relevance to patient care, if any.
Probiotic therapies and prophylactics may also confer some advantages for managing symptoms of COVID-19 or risks associated with its treatment.
Probiotics have tentatively been associated with the reduction of risk and duration of viral upper respiratory tract infections (1953–1955).
Some meta-analyses that have assessed the efficacy of probiotics in viral respiratory infections have reported moderate reductions in the incidence and duration of infection (1954, 1956).
Indeed, randomized controlled trials have shown that administering Bacillus subtilis and Enterococcus faecalis(1957), Lactobacillus rhamnosus GG(1958), or Lactobacillus casei and Bifidobacterium breve with galactooligosaccharides (1959) via the nasogastric tube to ventilated patients reduced the occurrence of ventilator-associated pneumonia in comparison to the respective control groups in studies of viral infections and sepsis.
These findings were also supported by a recent meta-analysis (1960).
Additionally, COVID-19 patients carry a significant risk of ventilator-associated bacterial pneumonia (1961), but it can be challenging for clinicians to diagnose this infection due to the fact that severe COVID-19 infection presents with the symptoms of pneumonia (1962).
Therefore, an effective prophylactic therapy for ventilator-associated pneumonia in severe COVID-19 patients would carry significant therapeutic value.
Additionally, in recent years, probiotics have become almost synonymous with the treatment of gastrointestinal issues due to their supposed anti-inflammatory and immunomodulatory effects (1963).
Notably, gastrointestinal symptoms commonly occur in COVID-19 patients (1964), and angiotensin-converting enzyme 2, the portal by which SARS-CoV-2 enters human cells, is highly expressed in enterocytes of the ileum and colon, suggesting that these organs may be a potential route of infection (1965, 1966).
Indeed, SARS-CoV-2 viral RNA has been detected in human feces (79, 540), and fecal-oral transmission of the virus has not yet been ruled out (1967).
Rectal swabs of some SARS-CoV-2 positive pediatric patients persistently tested positive for several days despite negative nasopharyngeal tests, indicating the potential for fecal viral shedding (1968).
However, there is conflicting evidence for the therapeutic value of various probiotics against the incidence or severity of gastrointestinal symptoms in viral or bacterial infections such as gastroenteritis (1969, 1970).
Nevertheless, it has been proposed that the administration of probiotics to COVID-19 patients and healthcare workers may prevent or ameliorate the gastrointestinal symptoms of COVID-19, a hypothesis that several clinical trials are now preparing to investigate (1971, 1972).
Other studies are investigating whether probiotics may affect patient outcomes following SARS-CoV-2 infection (1973).
Generally, the efficacy of probiotic use is a controversial topic among scientists.
In Europe, EFSA has banned the term probiotics on products labels, which has elicited either criticism for EFSA or support for probiotics from researchers in the field (1930, 1974, 1975).
This regulation is due to the hyperbolic claims placed on the labels of various probiotic products, which lack rigorous scientific data to support their efficacy.
Overall, the data supporting probiotics in the treatment or prevention of many different disorders and diseases is not conclusive, as the quality of the evidence is generally considered low (1953).
However, in the case of probiotics and respiratory infections, the evidence seems to be supportive of their potential therapeutic value.
Consequently, several investigations are underway to investigate the prophylactic and therapeutic potential of probiotics for COVID-19.
The blind use of conventional probiotics for COVID-19 is currently cautioned against until the pathogenesis of SARS-CoV-2 can be further established (1976).
Until clinical trials investigating the prophylactic and therapeutic potential of probiotics for COVID-19 are complete, it is not possible to provide an evidence-based recommendation for their use.
Despite these concerns, complementary use of probiotics as an adjuvant therapeutic has been proposed by the Chinese National Health Commission and National Administration of Traditional Chinese Medicine (80).
While supply issues prevented the probiotics market from showing the same rapid response to the COVID-19 as some other supplements, many suppliers are reporting growth during the pandemic (1977).
Therefore, the public response once again seems to have adopted supplements promoted as bolstering the immune response despite a lack of evidence suggesting they are beneficial for preventing or mitigating COVID-19.
10.9 Discussion
In this review, we report the findings to date of analyses of several dietary supplements and nutraceuticals.
While existing evidence suggests potential benefits of n-3 PUFA and probiotic supplementation for COVID-19 treatment and prophylaxis, clinical data is still lacking, although trials are underway.
Both zinc and vitamin C supplementation in hospitalized patients seem to be associated with positive outcomes; however, further clinical trials are required.
In any case, vitamin C and zinc intake are part of a healthy diet and likely present minimal risk when supplemented, though their potential prophylactic or therapeutic effects against COVID-19 are yet to be determined.
On the other hand, mounting evidence from observational studies indicates that there is an association between vitamin D deficiency and COVID-19 incidence has also been supported by meta-analysis (1978).
Indeed, scientists are working to confirm these findings and to determine whether a patient’s serum 25-hydroxyvitamin D levels are also associated with COVID-19 severity.
Clinical trials are required to determine whether preemptive vitamin D supplementation may mitigate against severe COVID-19.
In terms of the therapeutic potential of vitamin D, initial evidence from clinical trials is conflicting but seems to indicate that vitamin D supplementation may reduce COVID-19 severity (1909).
The various clinical trials currently underway will be imperative to provide information on the efficacious use of vitamin D supplementation for COVID-19 prevention and/or treatment.
The purported prophylactic and therapeutic benefits of dietary supplements and nutraceuticals for multiple disorders, diseases, and infections has been the subject of significant research and debate for the last few decades.
Inevitably, scientists are also investigating the potential for these various products to treat or prevent COVID-19.
This interest also extends to consumers, which led to a remarkable increase of sales of dietary supplements and nutraceuticals throughout the pandemic due to a desire to obtain additional protections from infection and disease.
The nutraceuticals discussed in this review, namely vitamin C, vitamin D, n-3 PUFA, zinc, and probiotics, were selected because of potential biological mechanisms that could beneficially affect viral and respiratory infections and because they are currently under clinical investigation.
Specifically, these compounds have all been found to influence cellular processes related to inflammation.
Inflammation is particularly relevant to COVID-19 because of the negative outcomes (often death) observed in a large number of patients whose immune response becomes hyperactive in response to SARS-CoV-2, leading to severe outcomes such as ARDS and sepsis (744).
Additionally, there is a well-established link between diet and inflammation (1979), potentially mediated in part by the microbiome (1980).
Thus, the idea that dietary modifications or supplementation could be used to modify the inflammatory response is tied to a broader view of how diet and the immune system are interconnected.
The supplements and nutraceuticals discussed here therefore lie in sharp contrast to other alleged nutraceutical or dietary supplements that have attracted during the pandemic, such as colloidal silver (1981), which have no known nutritional function and can be harmful.
Importantly, while little clinical evidence is available about the effects of any supplements against COVID-19, the risks associated with those discussed above are likely to be low, and in some cases, they can be obtained from dietary sources alone.
There are various other products and molecules that have garnered scientific interest and could merit further investigation.
These include polyphenols, lipid extracts, and tomato-based nutraceuticals, all of which have been suggested for the potential prevention of cardiovascular complications of COVID-19 such as thrombosis (1787, 1822).
Melatonin is another supplement that has been identified as a potential antiviral agent against SARS-CoV-2 using computational methods (1982), and it has also been highlighted as a potential therapeutic agent for COVID-19 due to its documented antioxidant, anti-apoptotic, immunomodulatory, and anti-inflammatory effects (1822, 1983, 1984).
Notably, melatonin, vitamin D and zinc have attracted public attention because they were included in the treatment plan of the former President of the United States upon his hospitalization due to COVID-19 (1985).
These are just some of the many substances and supplements that are currently under investigation but as of yet lack evidence to support their use for the prevention or treatment of COVID-19.
While there is plenty of skepticism put forward by physicians and scientists surrounding the use of supplements, these statements have not stopped consumers from purchasing these products, with one study reporting that online searches for dietary supplements in Poland began trending with the start of the pandemic (1928).
Additionally, supplement usage increased between the first and second wave of the pandemic.
Participants reported various reasons for their use of supplements, including to improve immunity (60%), to improve overall health (57%), and to fill nutrient gaps in their diet (53%).
Other efforts to collect large datasets regarding such behavior have also sought to explore a possible association between vitamin or supplement consumption and COVID-19.
An observational analysis of survey responses from 327,720 users of the COVID Symptom Study App found that the consumption of n-3 PUFA supplements, probiotics, multivitamins, and vitamin D was associated with a lower risk of SARS-CoV-2 infection in women but not men after adjusting for potential confounders (1986).
According to the authors, the sexual dimorphism observed may in part be because supplements may better support females due to known differences between the male and female immune systems, or it could be due to behavioral and health consciousness differences between the sexes (1986).
Certainly, randomized controlled trials are required to investigate these findings further.
Finally, it is known that a patient’s nutritional status affects health outcomes in various infectious diseases (1761), and COVID-19 is no different (1759, 1987, 1988).
Some of the main risk factors for severe COVID-19, which also happen to be linked to poor nutritional status, include obesity, hypertension, cardiovascular diseases, type II diabetes mellitus, and indeed age-related malnutrition (1757, 1759, 1989).
Although not the main focus of this review, it is important to consider the nutritional challenges associated with severe COVID-19 patients.
Hospitalized COVID-19 patients tend to report an unusually high loss of appetite preceding admission, some suffer diarrhea and gastrointestinal symptoms that result in significantly lower food intake, and patients with poorer nutritional status were more likely to have worse outcomes and require nutrition therapy (1990).
Dysphagia also seems to be a significant problem in pediatric patients that suffered multisystem inflammatory syndrome (1991) and rehabilitating COVID-19 patients, potentially contributing to poor nutritional status (1992).
Almost two-thirds of discharged COVID-19 ICU patients exhibit significant weight loss, of which 26% had weight loss greater than 10% (1988).
As investigated in this review, hospitalized patients also tend to exhibit vitamin D deficiency or insufficiency, which may be associated with greater disease severity (1978).
Therefore, further research is required to determine how dietary supplements and nutraceuticals may contribute to the treatment of severely ill and rehabilitating patients, who often rely on enteral nutrition.
10.10 Conclusions
Despite all the potential benefits of nutraceutical and dietary supplement interventions presented, currently there is a paucity of clinical evidence to support their use for the prevention or mitigation of COVID-19 infection.
Nevertheless, optimal nutritional status can prime an individual’s immune system to protect against the effects of acute respiratory viral infections by supporting normal maintenance of the immune system (1757, 1761).
Nutritional strategies can also play a role in the treatment of hospitalized patients, as malnutrition is a risk to COVID-19 patients (1992).
Overall, supplementation of vitamin C, vitamin D, and zinc may be an effective method of ensuring their adequate intake to maintain optimal immune function, which may also convey beneficial effects against viral infections due to their immunomodulatory effects.
Individuals should pay attention to their nutritional status, particularly their intake of vitamin D, considering that vitamin D deficiency is widespread.
The prevailing evidence seems to indicate an association between vitamin D deficiency with COVID-19 incidence and, potentially, severity (1884).
As a result, some international authorities have advised the general public, particularly those at high risk of infection, to consider vitamin D supplementation.
However, further well-controlled clinical trials are required to confirm these observations.
Many supplements and nutraceuticals designed for various ailments that are available in the United States and beyond are not strictly regulated (1993).
Consequently, there can be safety and efficacy concerns associated with many of these products.
Often, the vulnerable members of society can be exploited in this regard and, unfortunately, the COVID-19 pandemic has proven no different.
As mentioned above, the FDA has issued warnings to several companies for advertising falsified claims in relation to the preventative and therapeutic capabilities of their products against COVID-19 (1994).
Further intensive investigation is required to establish the effects of these nutraceuticals, if any, against COVID-19.
Until more effective therapeutics are established, the most effective mitigation strategies consist of encouraging standard public health practices such as regular hand washing with soap, wearing a face mask, and covering a cough with your elbow (1995), along with following social distancing measures, “stay at home” guidelines, expansive testing, and contact tracing (1996, 1997).
Indeed, in light of this review, it would also be pertinent to adopt a healthy diet and lifestyle following national guidelines in order to maintain optimal immune health.
Because of the broad public appeal of dietary supplements and nutraceuticals, it is important to evaluate the evidence regarding the use of such products.
We will continue to update this review as more findings become available.
11 Social Factors Influencing COVID-19 Exposure and Outcomes
11.1 Social Factors Influencing COVID-19 Outcomes
In addition to understanding the fundamental biology of the SARS-CoV-2 virus and COVID-19, it is critical to consider how the broader environment can influence both COVID-19 outcomes and efforts to develop and implement treatments for the disease.
The evidence clearly indicates that social environmental factors are critical determinants of individuals’ and communities’ risks related to COVID-19.
There are distinct components to COVID-19 susceptibility, and an individual’s risk can be elevated at one or all stages from exposure to recovery/mortality: an individual may be more likely to be exposed to the virus, more likely to get infected once exposed, more likely to have serious complications once infected, and be less likely to receive adequate care once they are seriously ill.
The fact that differences in survival between Black and white patients were no longer significant after controlling for comorbidities and socioeconomic status (type of insurance, neighborhood deprivation score, and hospital where treatment was received) in addition to sex and age (1998) underscores the relevance of social factors to understanding mortality differences between racial and ethnic groups.
Moreover, the Black patients were younger and more likely to be female than white patients, yet still had a higher mortality rate without correction for the other variables (1998).
Here, we outline a few systemic reasons that may exacerbate the COVID-19 pandemic in communities of color.
11.2 Factors Observed to be Associated with Susceptibility
As COVID-19 has spread into communities around the globe, it has become clear that the risks associated with this disease are not equally shared by all individuals or all communities.
Significant disparities in outcomes have led to interest in the demographic, biomedical, and social factors that influence COVID-19 severity.
Untangling the factors influencing COVID-19 susceptibility is a complex undertaking.
Among patients who are admitted to the hospital, outcomes have generally been poor, with rates of admission to the intensive care unit (ICU) upwards of 15% in both Wuhan, China and Italy (82, 1999, 2000).
However, hospitalization rates vary by location (2001).
This variation may be influenced by demographic (e.g., average age in the area), medical (e.g., the prevalence of comorbid conditions such as diabetes), and social (e.g., income or healthcare availability) factors that vary geographically.
Additionally, some of the same factors may influence an individual’s probability of exposure to SARS-CoV-2, their risk of developing a more serious case of COVID-19 that would require hospitalization, and their access to medical support.
As a result, quantifying or comparing susceptibility among individuals, communities, or other groups requires consideration of a number of complex phenomena that intersect across many disciplines of research.
In this section, the term “risk factor” is used to refer to variables that are statistically associated with more severe COVID-19 outcomes.
Some are intrinsic characteristics that have been observed to carry an association with variation in outcomes, whereas others may be more functionally linked to the pathophysiology of COVID-19.
11.2.1 Patient Traits Associated with Increased Risk
Two traits that have been consistently associated with more severe COVID-19 outcomes are male sex and advanced age (typically defined as 60 or older, with the greatest risk among those 85 and older (2002)).
In the United States, males and older individuals diagnosed with COVID-19 were found to be more likely to require hospitalization (2003, 2004).
A retrospective study of hospitalized Chinese patients (83) found that a higher probability of mortality was associated with older age, and world-wide, population age structure has been found to be an important variable for explaining differences in outbreak severity (2005).
The CFR for adults over 80 has been estimated upwards of 14% or even 20% (2006).
Male sex has also been identified as a risk factor for severe COVID-19 outcomes, including death (2007, 2008, 2009/).
Early reports from China and Europe indicated that even though the case rates were similar across males and females, males were at elevated risk for hospital admission, ICU admission, and death (2008), although data from some US states indicates more cases among females, potentially due to gender representation in care-taking professions (2010).
In older age groups (e.g., age 60 and older), comparable absolute numbers of male and female cases actually suggests a higher rate of occurrence in males, due to increased skew in the sex ratio (2008).
Current estimates based on worldwide data suggest that, compared to females, males may be 30% more likely to be hospitalized, 80% more likely to be admitted to the ICU, and 40% more likely to die as a result of COVID-19 (2009/).
There also may be a compounding effect of advanced age and male sex, with differences time to recovery worst for males over 60 years old relative to female members of their age cohort (2011).
Both of these risk factors can be approached through the lens of biology.
The biological basis for greater susceptibility with age is likely linked to the prevalence of extenuating health conditions such as heart failure or diabetes (2006).
Several hypotheses have been proposed to account for differences in severity between males and females.
For example, some evidence suggests that female sex hormones may be protective (2008, 2010).
ACE2 expression in the kidneys of male mice was observed to be twice as high as that of females, and a regulatory effect of estradiol on ACE2 expression was demonstrated by removing the gonads and then supplementing with estradiol (2010, 2012).
Other work in mice has shown an inverse association between mortality due to SARS-CoV-1 and estradiol, suggesting a protective role for the sex hormone (2010).
Similarly, evidence suggests that similar patterns might be found in other tissues.
A preliminary analysis identified higher levels of ACE2 expression in the myocardium of male patients with aortic valve stenosis showed than female patients, although this pattern was not found in controls (2008).
Additionally, research has indicated that females respond to lower doses than males of heart medications that act on the Renin angiotensin aldosterone system (RAAS) pathway, which is shared with ACE2 (2008).
Additionally, several components of the immune response, including the inflammatory response, may differ in intensity and timing between males and females (2010, 2012).
This hypothesis is supported by some preliminary evidence showing that female patients who recovered from severe COVID-19 had higher antibody titers than males (2010).
Sex steroids can also bind to immune cell receptors to influence cytokine production (2008).
Additionally, social factors may influence risks related to both age and sex: for example, older adults are more likely to live in care facilities, which have been a source for a large number of outbreaks (2013), and gender roles may also influence exposure and/or susceptibility due to differences in care-taking and/or risky behaviors (e.g., caring for elder relatives and smoking, respectively) (2008) among men and women (however, it should be noted that both transgender men and women are suspected to be at heightened risk (2014).)
11.2.2 Comorbid Health Conditions
A number of pre-existing or comorbid conditions have repeatedly been identified as risk factors for more severe COVID-19 outcomes.
Several underlying health conditions were identified at high prevalence among hospitalized patients, including obesity, diabetes, hypertension, lung disease, and cardiovascular disease (2001).
Higher Sequential Organ Failure Assessment (SOFA) scores have been associated with a higher probability of mortality (83), and comorbid conditions such as cardiovascular and lung disease as well as obesity were also associated with an increased risk of hospitalization and death, even when correcting for age and sex (2007).
Diabetes may increase the risk of lengthy hospitalization (2015) or of death (2015, 2016).
(2017) and (2018) discuss possible ways in which COVID-19 and diabetes may interact.
Obesity also appears to be associated with higher risk of severe outcomes from SARS-CoV-2 (2019, 2020).
Obesity is considered an underlying risk factor for other health problems, and the mechanism for its contributions to COVID-19 hospitalization or mortality is not yet clear (2021).
Dementia and cancer were also associated with the risk of death in an analysis of a large number (more than 20,000) COVID-19 patients in the United Kingdom (2007).
It should be noted that comorbid conditions are inextricably tied to age, as conditions tend to be accumulated over time, but that the prevalence of individual comorbidities or of population health overall can vary regionally (2022).
Several comorbidities that are highly prevalent in older adults, such as COPD, hypertension, cardiovascular disease, and diabetes, have been associated with CFRs upwards of 8% compared to an estimate of 1.4% in people without comorbidities (2006, 2023).
Therefore, both age and health are important considerations when predicting the impact of COVID-19 on a population (2022).
However, other associations may exist, such as patients with sepsis having higher SOFA scores – in fact, SOFA was developed for the assessment of organ failure in the context of sepsis, and the acronym originally stood for Sepsis-Related Organ Failure Assessment (2024, 2025).
Additionally, certain conditions are likely to be more prevalent under or exacerbated by social conditions, especially poverty, as is discussed further below.
11.2.3 Ancestry
A number of studies have suggested associations between individuals’ racial and ethnic backgrounds and their COVID-19 risk.
In particular, Black Americans are consistently identified as carrying a higher burden of COVID-19 than white Americans (2003, 2004), with differences in the rates of kidney complications from COVID-19 particularly pronounced (89).
Statistics from a number of cities indicate significant discrepancies between the proportion of COVID-19 cases and deaths in Black Americans relative to their representation in the general population (2026).
In addition to Black Americans, disproportionate harm and mortality from COVID-19 has also been noted in Latino/Hispanic Americans and in Native American and Alaskan Native communities, including the Navajo nation [(2027); (2028); (2029); https://www.nytimes.com/2020/04/09/us/coronavirus-navajo-nation.html?searchResultPosition=10; (2030); (2031); (2032)].
In Brazil, indigenous communities likewise carry an increased burden of COVID-19 (2033).
In the United Kingdom, nonwhite ethnicity (principally Black or South Asian) was one of several factors found to be associated with a higher risk of death from COVID-19 (2034).
From a genetic standpoint, it is highly unlikely that ancestry itself predisposes individuals to contracting COVID-19 or to experiencing severe COVID-19 outcomes.
Examining human genetic diversity indicates variation over a geographic continuum, and that most human genetic variation is associated with the African continent (2035).
African-Americans are also a more genetically diverse group relative to European-Americans, with a large number of rare alleles and a much smaller fraction of common alleles identified in African-Americans (2036).
Therefore, the idea that African ancestry (at the continent level) might convey some sort of genetic risk for severe COVID-19 contrasts with what is known about worldwide human genetic diversity (2037).
The possibility for genetic variants that confer some risk or some protection remains possible, but has not been widely explored, especially at a global level.
Research in Beijing of a small number (n=80) hospitalized COVID-19 patients revealed an association between severe COVID-19 outcomes and homozygosity for an allele in the interferon-induced transmembrane protein 3 (IFITM3) gene, which was selected as a candidate because it was previously found to be associated with influenza outcomes in Chinese patients (435).
Genetic factors may also play a role in the risk of respiratory failure for COVID-19 (464, 2038, 2039).
However, genetic variants associated with outcomes within ancestral groups are far less surprising than genetic variants explaining outcomes between groups.
Alleles in ACE2 and TMPRSS2 have been identified that vary in frequency among ancestral groups (2040), but whether these variants are associated with COVID-19 susceptibility has not been explored.
Instead, examining patterns of COVID-19 susceptibility on a global scale that suggest that social factors are of primary importance in predicting mortality.
Reports from several sub-Saharan African countries have indicated that the effects of the COVID-19 pandemic have been less severe than expected based on the outbreaks in China and Italy.
In Kenya, for example, estimates of national prevalence based on testing blood donors for SARS-CoV-2 antibodies were consistent with 5% of Kenyan adults having recovered from COVID-19 (2041).
This high seroprevalence of antibodies lies in sharp contrast to the low number of COVID-19 fatalities in Kenya, which at the time was 71 out of 2093 known cases (2041).
Likewise, a serosurvey of health care workers in Blantyre City, Malawi reported an adjusted antibody prevalence of 12.3%, suggesting that the virus had been circulating more widely than thought and that the death rate was up eight times lower than models had predicted (2042).
While several possible hypotheses for the apparent reduced impact of COVID-19 on the African continent are being explored, such as young demographics in many places (2043), these reports present a stark contrast to the severity of COVID-19 in Americans and Europeans of African descent.
Additionally, ethnic minorities in the United Kingdom also tend to be younger than white British living in the same areas, yet the burden of COVID-19 is still more serious for minorities, especially people of Black Caribbean ancestry, both in absolute numbers and when controlling for age and location (2044/).
Furthermore, the groups in the United States and United Kingdom that have been identified as carrying elevated COVID-19 burden, namely Black American, indigenous American, and Black and South Asian British, are quite distinct in their position on the human ancestral tree.
What is shared across these groups is instead a history of disenfranchisement under colonialism and ongoing systematic racism.
A large analysis of over 11,000 COVID-19 patients hospitalized in 92 hospitals across U.S. states revealed that Black patients were younger, more often female, more likely to be on Medicaid, more likely to have comorbidities, and came from neighborhoods identified as more economically deprived than white patients (1998).
This study reported that when these factors were accounted for, the differences in mortality between Black and white patients were no longer significant.
Thus, the current evidence suggests that the apparent correlations between ancestry and health outcomes must be examined in the appropriate social context.
11.3 Environmental Influences on Susceptibility
11.3.1 Exposure to COVID-19
Social distancing has emerged as one of the main social policies used to manage the COVID-19 epidemic in many countries.
Many governments issued stay-at-home orders, especially in the initial months of the crisis.
However, data clearly indicates that these orders impacted different socioeconomic groups differently.
In U.S. counties with and without stay-at-home orders, smartphone tracking indicated a significant decrease in the general population’s mobility in April relative to February through March of 2020 (-52.3% and -60.8%, respectively) (2045).
A linear relationship was observed between counties’ reduction in mobility and their wealth and health, as measured by access to health care, food security, income, space, and other factors (2045).
Counties with greater reductions in mobility were also found to have much lower child poverty and household crowding and to be more racially segregated, and to have fewer youth and more elderly residents (2045).
Similar associations between wealth and decreased mobility were observed in cellphone GPS data from Colombia, Indonesia, and Mexico collected between January and May 2020 (2046), as well as in a very large data set from several US cities (2047).
These disparities in mobility are likely to be related to the role that essential workers have played during the pandemic.
Essential workers are disproportionately likely to be female, people of color, immigrants, and to have an income below 200% of the poverty line (2048).
Black Americans in particular are over-represented among front-line workers and in professions where social distancing is infeasible (2049).
Health care work in particular presents an increased risk of exposure to SARS-CoV-2 (2049–2053).
In the United Kingdom, (South) Asians are more likely than their white counterparts to be medical professionals (2044/), although BAME medical professionals are still disproportionately represented in the proportion of National Health Service staff deaths (2054).
Similar trends have been reported for nurses, especially nurses of color, in the United States (2055/-/files/graphics/0920_Covid19_SinsOfOmission_Data_Report.pdf).
Furthermore, beyond the risks associated with work itself, use of public transportation may also impact COVID-19 risk (2056).
The socioeconomic and racial/ethnic gaps in who is working on the front lines of the pandemic make it clear that socioeconomic privilege is likely to decrease the probability of exposure to SARS-CoV-2.
Increased risk of exposure can also arise outside the workplace.
Nursing homes and skilled nursing facilities received attention early on as high-risk locations for COVID-19 outbreaks (2057).
Prisons and detention centers also confer a high risk of exposure or infection (2058, 2059).
Populations in care facilities are largely older adults, and in the United States, incarcerated people are more likely to be male and persons of color, especially Black (2060).
Additionally, multi-generational households are less common among non-Hispanic white Americans than people of other racial and ethnic backgrounds (2061), increasing the risk of exposure for more susceptible family members.
Analysis suggests that household crowding may also be associated with increased risk of COVID-19 exposure (2045), and household crowding is associated with poverty (2062).
Forms of economic insecurity like housing insecurity, which is associated with poverty and more pronounced in communities subjected to racism (2063, 2064), would be likely to increase household crowding and other possible sources of exposure.
As a result, facets of systemic inequality such as mass incarceration of Black Americans and poverty are likely to increase the risk of exposure outside of the workplace.
11.3.2 Severity of COVID-19 Following Exposure
Following exposure to SARS-CoV-2, the likelihood that an individual develops COVID-19 and the severity of the disease presentation can be influenced by a number of social factors.
As discussed above, a number of patient characteristics are associated with the likelihood of severe COVID-19 symptoms.
In some cases, these trends run counter to those expected given rates of exposure: for example, although women are more likely to be exposed, men are more likely to be diagnosed with, hospitalized from, or die from COVID-19 (2010).
In the case of comorbid conditions and racial/ethnic demographics, however, social factors are highly likely to modulate or at least influence the apparent association between these traits and the increased risk from COVID-19.
In particular, the comorbidities and racial/ethnic correlates of severe COVID-19 outcomes suggest that poverty confers additional risk for COVID-19.
In order to explore the relationship between poverty and COVID-19 outcomes, it is necessary to consider how poverty impacts biology.
In particular, we focus on the United States and the United Kingdom.
Comorbidities that increase risk for COVID-19, including obesity, type II diabetes, hypertension, and cardiovascular disease, are known to be intercorrelated (2065).
Metabolic conditions related to heightened inflammation, like obesity, type II diabetes, and hypertension, are more strongly associated with negative COVID-19 outcomes than other comorbid conditions, such as chronic heart disease (2066).
As discussed above, dysregulated inflammation characteristic of cytokine release syndrome is one of the greatest concerns for COVID-19-related death.
Therefore, it is possible that chronic inflammation characteristic of these metabolic conditions predisposes patients to COVID-19-related death (2066).
The association between these diseases and severe COVID-19 outcomes is a concern from a health equity perspective because poverty exposes people to “obesogenic” conditions (2067) and is therefore unsurprisingly associated with higher incidence of obesity and associated disorders (2068).
Furthermore, cell phone GPS data suggests that lower socioeconomic status may also be associated with decreased access to healthy food choices during the COVID-19 pandemic (2069, 2070), suggesting that health-related risk factors for COVID-19 may be exacerbated as the pandemic continues (2071).
Chronic inflammation is a known outcome of chronic stress (e.g., (2072–2075)).
Therefore, the chronic stress of poverty is likely to influence health broadly (as summarized in (2076)) and especially during the stress of the ongoing pandemic.
A preprint (2077) provided observational evidence that geographical areas in the United States that suffer from worse air pollution by fine particulate matter have also suffered more COVID-19 deaths per capita, after adjusting for demographic covariates.
Although lack of individual-level exposure data and the impossibility of randomization make it difficult to elucidate the exact causal mechanism, this finding would be consistent with similar findings for all-cause mortality (e.g., (2078)).
Exposure to air pollution is associated with both poverty (e.g., (2079)) and chronic inflammation (2080).
Other outcomes of environmental racism, such as the proximity of abandoned uranium mines to Navajo land, can also cause respiratory illnesses and other health issues (2032).
Similarly, preliminary findings indicate that nutritional status (e.g., vitamin D deficiency (1893)) may be associated with COVID-19 outcomes, and reduced access to grocery stores and fresh food often co-occurs with environmental racism (2032, 2081).
Taken together, the evidence suggests that low-income workers who face greater exposure to SARS-CoV-2 due to their home or work conditions are also more likely to face environmental and social stressors associated with increased inflammation, and therefore with increased risk from COVID-19.
In particular, structural racism can play an important role on disease severity after SARS-CoV-2 exposure, due to consequences of racism which include an increased likelihood of poverty and its associated food and housing instability.
COVID-19 can thus be considered a “syndemic”, or a synergistic interaction between several epidemics (2082).
As a result, it is not surprising that people from minoritized backgrounds and/or with certain pre-existing conditions are more likely to suffer severe effects of COVID-19, but these “risk factors” are likely to be causally linked to poverty (2083).
11.3.3 Access to Treatment
Finally, COVID-19 outcomes can be influenced by access to healthcare.
Receiving care for COVID-19 can, but does not always, include receiving a positive test for the SARS-CoV-2 virus.
For example, it is common to see treatment guidelines for suspected cases regardless of whether the presence of SARS-CoV-2 has been confirmed (e.g., (2084)).
Whether and where a patient is diagnosed can depend on their access to testing, which can vary both between and within countries.
In the United States, it is not always clear whether an individual will have access to free testing (2085, 2086).
The concern has been raised that more economic privilege is likely to correspond to increased access to testing, at least within the United States (2087).
This is supported by the fact that African Americans seem to be more likely to be diagnosed in the hospital, while individuals from other groups were more likely to have been diagnosed in ambulatory settings in the community (2003).
Any delays in treatment are a cause for concern (2087), which could potentially be increased by an inability to acquire testing because in the United States, insurance coverage for care received can depend on a positive test (2088).
Another important question is whether patients with moderate to severe cases are able to access hospital facilities and treatments, to the extent that they have been identified.
Early findings from China as of February 2020 suggested the COVID-19 mortality rate to be much lower in the most developed regions of the country (2089), although reported mortality is generally an estimate of CFR, which is dependent on rates of testing.
Efforts to make treatment accessible for all confirmed and suspected cases of COVID-19 in China are credited with expanding care to people with fewer economic resources (2090).
In the United States, access to healthcare varies widely, with certain sectors of the workforce less likely to have health insurance; many essential workers in transportation, food service, and other frontline fields are among those likely to be uninsured or underinsured (2087).
As of 2018, Hispanic Americans of all races were much less likely to have health insurance than people from non-Hispanic backgrounds (2091).
Therefore, access to diagnostics and care prior to the development of severe COVID-19 is likely to vary depending on socioeconomic and social factors, many of which overlap with the risks of exposure and of developing more severe COVID-19 symptoms.
This discrepancy ties into concerns about broad infrastructural challenges imposed by COVID-19.
A major concern in many countries has been the saturation of healthcare systems due to the volume of COVID-19 hospitalizations (e.g., (320)).
Similarly, there have been shortages of supplies such as ventilators that are critical to the survival of many COVID-19 patients, leading to extensive ethical discussions about how to allocate limited resources among patients (2092–2095).
Although it is generally considered unethical to consider demographic factors such as age, sex, race, or ethnicity while making such decisions, and ideally this information would not be shared with triage teams tasked with allocating limited resources among patients (2096), there are substantial concerns about implicit and explicit biases against older adults (2097), premature infants (2098), and people with disabilities or comorbidities (2096, 2099, 2100).
Because of the greater burden of chronic disease in populations subjected to systemic racism, algorithms intended to be blind to race and ethnicity could, in fact, reinforce systemic inequalities caused by structural racism (2101–2103).
Because of this inequality, it has been argued that groups facing health disparities should be prioritized by these algorithms (2104).
This approach would carry its own ethical concerns, including the fact that many resources that need to be distributed do not have well-established risks and benefits (2104).
As the pandemic has progressed, it has become clear that ICU beds and ventilators are not the only limited resources that needs to be allocated, and, in fact, the survival rate for patients who receive mechanical ventilation is lower than these discussions would suggest (2105).
Allocation of interventions that may reduce suffering, including palliative care, has become critically important (2105, 2106).
The ambiguities surrounding the risks and benefits associated with therapeutics that have been approved under emergency use authorizations also present ethical concerns related to the distribution of resources (2104).
For example, remdesivir, discussed above, is currently available for the treatment of COVID-19 under compassionate use guidelines and through expanded access programs, and in many cases has been donated to hospitals by Gilead (2107, 2108).
Regulations guiding the distribution of drugs in situations like these typically do not address how to determine which patients receive them (2108).
Prioritizing marginalized groups for treatment with a drug like remdesivir would also be unethical because it would entail disproportionately exposing these groups to a therapeutic that may or not be beneficial (2104).
On the other hand, given that the drug is one of the most promising treatments available for many patients, using a framework that tacitly feeds into structural biases would also be unethical.
At present, the report prepared for the Director of the CDC by Ethics Subcommittee of the CDC fails to address the complexity of this ethical question given the state of structural racism in the United States, instead stating that “prioritizing individuals according to their chances for short-term survival also avoids ethically irrelevant considerations, such as race or socioeconomic status” (2109).
In many cases, experimental therapeutics are made available only through participation in clinical trials (2110).
However, given the history of medical trials abusing minority communities, especially Black Americans, there is a history of unequal representation in clinical trial enrollment (2110).
As a result, the standard practice of requiring enrollment in a clinical trial in order to receive experimental treatment may also reinforce patterns established by systemic racism.
11.3.4 Access to and Representation in Clinical Trials
Experimental treatments are often made available to patients primarily or even exclusively through clinical trials.
The advantage of this approach is that clinical trials are designed to collect rigorous data about the effects of a treatment on patients.
The disadvantage is that access to clinical trials is not equal among all people who suffer from a disease.
Two important considerations that can impact an individual’s access to clinical trials are geography and social perceptions of clinical trials.
For the first, the geographic distribution of trial recruitment efforts are typically bounded and can vary widely among difference locations, and for the second, the social context of medical interactions can impact strategies for and the success of outreach to different communities.
Differential access to clinical trials raises concerns because it introduces biases that can influence scientific and medical research on therapeutics and prophylactics broadly.
Concerns about bias in clinical trials need to address both trial recruitment and operation.
In the present crisis, such biases are particularly salient because COVID-19 is a disease of global concern.
Treatment is needed by people all over the world, and clinical research that characterizes treatment outcomes in a variety of populations is critically important.
Global representation in clinical trials is important to ensuring that experimental treatments are available equally to COVID-19 patients who may need them.
The advantage to a patient of participation in a clinical trial is that they may receive an experimental treatment they would not have been able to access otherwise.
The potential downsides of participation include that the efficacy and side effects of such treatments are often poorly characterized and that patients who enroll in clinical trials will in some cases run the risk of being assigned to a placebo condition where they do not receive the treatment but miss out on opportunities to receive other treatments.
The benefits and burdens of clinical trials therefore need to be weighed carefully to ensure that they don’t reinforce existing health disparities.
The WHO Director‐General Tedros Adhanom Ghebreyesus stated his condemnation of utilizing low and middle income countries as test subjects for clinical trials, yet having highly developed countries as the majority of clinical trial representation is also not the answer (2111).
Figure 13 showcases two choropleths detailing COVID-19 clinical trial recruitment by country.
China, the United States, and France are among the countries with the most clinical trial recruiting for trials with single-country enrollment.
Many countries have little to no clinical trial recruiting, with the continents of Africa and South America much less represented than Asia, Europe, and North America.
Trials that recruit across multiple countries do appear to broaden geographic representation, but these trials seem to be heavily dominated by the United States and European Union.
Figure 13:Geographic distribution of COVID-19 clinical trials.
The density of clinical trials is reported at the country level.
As of December 31, 2020, there are 6,987 trials in the University of Oxford Evidence-Based Medicine Data Lab’s COVID-19 TrialsTracker (739), of which 3,962 are interventional.
The top figure demonstrates the density of interventional trials recruiting only from a singular country, while the bottom shows the distribution of recruitment for interventional trials that involve more than one country.
A few different concerns arise from this skewed geographic representation in clinical trial recruitment.
First, treatments such as remdesivir that are promising but primarily available to clinical trial participants are unlikely to be accessible by people in many countries.
Second, it raises the concern that the findings of clinical trials will be based on participants from many of the wealthiest countries, which may lead to ambiguity in whether the findings can be extrapolated to COVID-19 patients elsewhere.
Especially with the global nature of COVID-19, equitable access to therapeutics and vaccines has been a concern at the forefront of many discussions about policy (e.g., (2112), yet data like that shown in Figure 13 demonstrates that accessibility is likely to be a significant issue.
Another concern with the heterogeneous international distribution of clinical trials is that the governments of countries leading these clinical trials might prioritize their own populations once vaccines are developed, causing unequal health outcomes (2113).
Additionally, even within a single state in the United States (Maryland), geography was found to influence the likelihood of being recruited into or enrolled in a clinical trial, with patients in under-served rural areas less likely to enroll (2114).
Thus, geography both on the global and local levels may influence when treatments and vaccines are available and who is able to access them.
Efforts such as the African Union’s efforts to coordinate and promote vaccine development (2115) are therefore critical to promoting equity in the COVID-19 response.
Even when patients are located within the geographic recruitment area of clinical trials, however, there can still be demographic inequalities in enrollment.
When efforts are made to ensure equal opportunity to participate in clinical trials, there is no significant difference in participation among racial/ethnic groups (2116).
However, within the United States, real clinical trial recruitment numbers have indicated for many years that racial minorities, especially African-Americans, tend to be under-represented (e.g., (2117–2120)).
This trend is especially concerning given the disproportionate impact of COVID-19 on African-Americans.
Early evidence suggests that the proportion of Black, Latinx, and Native American participants in clinical trials for drugs such as remdesivir is much lower than the representation of these groups among COVID-19 patients (2121).
One proposed explanation for differences among racial and ethnic groups in clinical trial enrollment refers to different experiences in healthcare settings.
While some plausible reasons for the disparity in communication between physicians and patients could be a lack of awareness and education, mistrust in healthcare professionals, and a lack of health insurance (2116), a major concern is that patients from certain racial and ethnic groups are marginalized even while seeking healthcare.
In the United States, many patients experience “othering” from physicians and other medical professionals due to their race or other external characteristics such as gender (e.g., (2122)).
Many studies have sought to characterize implicit biases in healthcare providers and whether they affect their perceptions or treatment of patients.
A systematic review that examined 37 such studies reported that most (31) identified racial and/or ethnic biases in healthcare providers in many different roles, although the evidence about whether these biases translated to different attitudes towards patients was mixed (2123), with similar findings reported by a second systematic review (2124).
However, data about real-world patient outcomes are very limited, with most studies relying on clinical vignette-based exercises (2123), and other analyses suggest that physician implicit bias could impact the patient’s perception of the negativity/positivity of the interaction regardless of the physician’s explicit behavior towards the patient (2125).
Because racism is a common factor in both, negative patient experiences with medical professionals are likely to compound other issues of systemic inequality, such as a lack of access to adequate care, a lack of insurance, or increased exposure to SARS-CoV-2 (2126).
Furthermore, the experience of being othered is not only expected to impact patients’ trust in and comfort with their provider, but also may directly impact whether or not the patient is offered the opportunity to participate in a clinical trial at all.
Some studies suggest communication between physicians and patients impacts whether or not a physician offers a patient participation in a clinical trial.
For example, researchers utilized a linguistic analysis to assess mean word count of phrases related to clinical trial enrollment, such as voluntary participation, clinical trial, etc. (2116).
The data indicated that the mean word count of the entire visit was 1.5 times more for white patients in comparison to Black patients.
In addition, the greatest disparity between white and Black patients’ experience was the discussion of risks, with over 2 times as many risk-related words spoken with white patients than Black patients (2116).
The trends observed for other clinical trials raise the concern that COVID-19 clinical trial information may not be discussed as thoroughly or as often with Black patients compared to white patients.
These discrepancies are especially concerning given that many COVID-19 treatments are being or are considered being made available to patients prior to FDA approval through Emergency Use Authorizations.
In the past, African-Americans have been over-represented relative to national demographics in use of the FDA’s Exception From Informed Consent (EFIC) pathway (2127).
Through this pathway, people who are incapacitated can receive an experimental treatment even if they are not able to consent and there is not sufficient time to seek approval from an authorized representative.
This pathway presents concerns, however, when it is considered in the context of a long history of systematic abuses in medical experimentation where informed consent was not obtained from people of color, such as the Tuskegee syphilis experiments (2128).
While the goal of EFIC approval is to provide treatment to patients who urgently need it, the combination of the ongoing legacy of racism in medicine renders this trend concerning.
With COVID-19, efforts to prioritize people who suffer from systemic racism are often designed with the goal of righting some of these inequalities (e.g., (2129)), but particular attention to informed consent will be imperative in ensuring these trials are ethical given that the benefits and risks of emerging treatments are still poorly characterized.
Making a substantial effort to run inclusive clinical trials is also important because of the possibility that racism could impact how a patient responds to a treatment.
For example, as discussed above, dexamethasone has been identified as a promising treatment for patients experiencing cytokine release syndrome, but the mechanism of action is tied to the stress response.
A study from 2005 reported that Black asthma patients showed reduced responsiveness to dexamethasone in comparison to white patients and suggested Black patients might therefore require higher doses of the drug (2130).
In the context of chronic stress caused by systemic racism, this result is not surprising: chronic stress is associated with dysregulated production of glucocorticoids (2131) and glucocorticoid receptor resistance (2132).
However, it underscores the critical need for treatment guidelines to take into account differences in life experience, which would be facilitated by the recruitment of patients from a wide range of backgrounds.
Attention to the social aspects of clinical trial enrollment must therefore be an essential component of the medical research community’s response to COVID-19.
11.4 Conclusions and Future Directions
As the COVID-19 pandemic evolves, the scientific community’s response will be critical for identifying potential pharmacological and biotechnological developments that may aid in combating the virus and the disease it causes.
However, this global crisis highlights the importance of mounting a response based on collaboration among a wide variety of disciplines.
Understanding the basic science of the virus and its pathogenesis is imperative for identifying and envisioning possible diagnostic and therapeutic approaches; understanding how social factors can influence outcomes and shape implementation of a response is critical to disseminating any scientific advancements.
Summarizing such a complex and ever-changing topic presents a number of challenges.
This review represents the effort of over 50 contributors to distill and interpret the available information.
However, this text represents a dynamic and evolving document, and we welcome continued contributions from all researchers who have insights into how these topics intersect.
A multidisciplinary perspective is critical to understanding this evolving crisis, and in this review we seek to use open science tools to coordinate a response among a variety of researchers.
We intend to publish additional updates as the situation evolves.
12 An Open-Publishing Response to the COVID-19 Infodemic
12.1 ABSTRACT
The COVID-19 pandemic catalyzed the rapid dissemination of papers and preprints investigating the disease and its associated virus, SARS-CoV-2.
The multifaceted nature of COVID-19 demands a multidisciplinary approach, but the urgency of the crisis combined with the need for social distancing measures present unique challenges to collaborative science.
We applied a massive online open publishing approach to this problem using Manubot.
Through GitHub, collaborators summarized and critiqued COVID-19 literature, creating a review manuscript.
Manubot automatically compiled citation information for referenced preprints, journal publications, websites, and clinical trials.
Continuous integration workflows retrieved up-to-date data from online sources nightly, regenerating some of the manuscript’s figures and statistics.
Manubot rendered the manuscript into PDF, HTML, LaTeX, and DOCX outputs, immediately updating the version available online upon the integration of new content.
Through this effort, we organized over 50 scientists from a range of backgrounds who evaluated over 1,500 sources and developed seven literature reviews.
While many efforts from the computational community have focused on mining COVID-19 literature, our project illustrates the power of open publishing to organize both technical and non-technical scientists to aggregate and disseminate information in response to an evolving crisis.
KEYWORDS
COVID-19, living document, open publishing, open-source, data integration, manubot
12.2 INTRODUCTION
Coronavirus Disease 2019 (COVID-19) caused a worldwide public health crisis that has reshaped many aspects of society.
The scientific community has, in turn, devoted significant attention and resources towards COVID-19 and the associated virus, SARS-CoV-2, resulting in the release of data and publications at a rate and scale never previously seen for a single topic.
Over 20,000 articles about COVID-19 were released in the first four months of the pandemic (2133), causing an “infodemic” (2133, 2134).
The COVID-19 Open Research Dataset (CORD-19) (2135), which was developed in part with the goal of training machine learning algorithms on COVID-19-related text, illustrates the growth of related scholarly literature (Figure 14).
This resource was developed by querying several sources for terms related to SARS-CoV-2 and COVID-19, as well as the coronaviruses SARS-CoV-1 and MERS-CoV and their associated diseases (2135).
CORD-19 contained 1056660 manuscripts as of 2022-06-02.
Additional curation by CoronaCentral (2136) has produced, at present, a set of over 180,000 publications particularly relevant to COVID-19 and closely related viruses.
Despite many advances in understanding the virus and the disease, there are also downsides to the availability of so much information.
“Excessive publication” has been recognized as a concern for over forty years (2137) and has been discussed with respect to the COVID-19 literature (992).
Any effort to synthesize, summarize, and contextualize COVID-19 research will face a vast corpus of potentially relevant material.
Figure 14:Growth of the CORD-19 dataset.
The number of articles has proliferated, with both traditional and preprint manuscripts in the corpus.
The first release (March 16, 2020) contained 28,000 documents (2135).
As of 2022-06-02, this had increased to 1056660 articles.
Of these, 42343 are preprints from arXiv, medRxiv, and bioRxiv.
Information was released rapidly by both traditional publishers and preprint servers, and many papers faced subsequent scrutiny.
The number of COVID-19 papers retracted may be higher, and potentially much higher, than is typical, although a thorough investigation of this question requires more time to elapse (2138, 2139).
Many papers and preprints are also associated with corrections or expressions of concern1(2139).
Preprints are released prior to peer review, but some traditional publishing venues have fast-tracked COVID-19 papers through peer review, leading to questions about whether they are held to typical standards (2140).
Therefore, evaluating the COVID-19 literature requires not only digesting available information but also monitoring subsequent changes.
Because of the fast-moving nature of the topic, many efforts to summarize and synthesize the COVID-19 literature have been undertaken.
These efforts include newsletters2(2141), web portals3(2142) or the now-defunct http://covidpreprints.com4, comments on preprint servers5(2143), and even a journal6.
However, the explosive rate of publication presents challenges for such efforts, many of which are no longer active.
Similarly, many literature reviews have been written on the available COVID-19 literature (2144–2148), but static reviews quickly become outdated as new research is released or existing research is retracted or superseded.
One example is a review of topics in COVID-19 research including vaccine development (2148).
This review was published on July 10, 2020, four days before Moderna released the surprisingly promising results of their phase 1 trial (1119) that changed expectations surrounding vaccines.
Therefore, the COVID-19 publishing climate presented a challenge where curation of the literature by a diverse group of experts in a format that could respond quickly to high-volume, high-velocity information was desirable.
We therefore sought to develop a platform for scientific discussion and collaboration around COVID-19 by adapting open publishing infrastructure to accommodate the scale of COVID-19 publishing.
Recent advances in open publishing have created an infrastructure that facilitates distributed, version-controlled collaboration on manuscripts (2149).
Manubot (2149) is a collaborative framework developed to adapt open-source software development techniques and version control for manuscript writing.
With Manubot, manuscripts are managed and maintained using GitHub, a popular, online version control interface.
We selected Manubot because it offers several advantages over comparable collaborative writing platforms such as Authorea, Overleaf, Google Docs, Word Online, or wikis (2149).
Citation-by-identifier ensures consistent reference metadata standards that would be difficult to maintain manually in a manuscript with dozens of authors and over 1,500 citations.
Manubot’s pull request-based contribution model balances the goals of making the project open to everyone and maintaining scientific accuracy.
All contributions are reviewed, discussed, and formally approved on GitHub before text updates appear in the public-facing manuscript7.
Continuous integration (CI) seamlessly combines author-produced text and figures with automatically generated and updated statistics and figures derived from external data sources and the manuscript’s own content.
In addition, the authors who initially launched this project included Manubot developers who had prior successes using Manubot for massively open and traditional manuscripts.
Collaboration via massively open online papers has been identified as a strategy for promoting inclusion and interdisciplinary thought (2150).
However, the Manubot workflow can be intimidating to contributors who are not well-versed in git (2150).
The synthesis and discussion of the emerging literature by biomedical scientists and clinicians is imperative to a robust interpretation of COVID-19 research.
Such efforts in biology often rely on What You See Is What You Get tools such as Google Docs, despite the significant limitations of these platforms in the face of excessive publication.
We recognized that the problem of synthesizing the COVID-19 literature lent itself well to the Manubot platform, but that the potential technical expertise required to work with Manubot presented a barrier to domain experts.
Here, we describe the adaptation of Manubot to facilitate collaboration in the extreme case of the COVID-19 infodemic, with the objective of developing a centralized platform for summarizing and synthesizing a massive amount of preprints, news stories, journal publications, and data.
Unlike prior collaborations built on Manubot, most contributors to the COVID-19 collaborative literature review came from biology or medicine.
The members of the COVID-19 Review Consortium consolidated information about the virus in the context of related viruses and to synthesize rapidly emerging literature.
Manubot provided the infrastructure to manage contributions from the community and create a living, scholarly document integrating data from multiple sources.
Its back-end allowed biomedical scientists to sort and distill informative content out of the overwhelming flood of information (2151) in order to provide a resource that would be useful to the broader scientific community.
This case study demonstrates the value of open collaborative writing tools such as Manubot to emerging challenges.
Because it is open source software, we were able to adapt and customize Manubot to flexibly meet the needs of COVID-19 review.
Recording the evolution of information over time and assembling a resource that auto-updated in response to the evolving crisis revealed the particular value that Manubot holds for managing rapid changes in scientific thought.
12.3 METHODS
12.3.1 Contributor Recruitment and Roles
First, it was necessary to establish Manubot as a platform accessible to researchers with limited experience working version control, given that this is not typically emphasized in biology and medicine (2152–2154).
Contributors were recruited primarily by word of mouth and on Twitter, and we also collaborated with existing efforts to train early-career researchers.
We invited potential collaborators to contribute a short introduction on a GitHub issue in order to collect information about participants and provide an introduction to working with GitHub issues.
Interested participants were encouraged to contribute in several ways.
One option was to catalog articles of interest as issues.
We developed a standardized set of questions for contributors to consider when evaluating an article following a framework often used for assessing medical literature.
This approach emphasizes examining the methods used, assignment (whether the study was observational or randomized), assessment, results, interpretation, and how well the study extrapolates (2155).
Contributors were also invited to contribute or edit text using GitHub’s pull request system.
These contributions were not strictly defined and could range from minor corrections to punctuation and grammar to large-scale additions of text.
Finally, a small number of contributors (the authors of this paper) contributed technical expertise, either through the development of standardized approaches to the evaluation of papers based on the MAARIE Framework (2156), the writing of code to generate manuscript figures, or the addition of features to Manubot.
All of these additions were also submitted as pull requests, either to the COVID-19 review repository or to an external repository, as appropriate.
Each pull request was reviewed and approved by at least one other contributor before being merged into the main branch.
We tagged potential reviewers based on the introductions they had contributed in order to encourage participation.
Authorship was determined based on the Contributor Roles Taxonomy8.
Due to the permeability of ideas among different sections, contributors to a specific manuscript were recognized with masthead authorship, while all contributors to the project were recognized with consortium authorship on all papers.
Emphasizing the use of issues and pull requests was designed to encourage authors with and without git experience to discuss papers and provide feedback (both formal and informal) on proposed text additions or changes.
We also used the Gitter chat platform9 to promote informal questions and sharing of information among collaborators.
12.3.2 Utilization and Expansion of Manubot
Applying Manubot’s existing capabilities allowed us to confront several challenges common in large-scale collaborations, such as maintaining a record of contributions that allowed us to allocate credit appropriately or to contact the original author if questions arose.
Additionally, an up-to-date version of the content was available at all times online in HTML10 or PDF format11.
This approach also allowed us to minimize the demand on authors to curate and sync bibliographic resources.
Manubot provides the functionality to create a bibliography using digital object identifiers (DOIs), website URLs, or other identifiers such as PubMed identifiers and arXiv IDs.
The author can insert a citation in-line using a format such as [@doi:10.1371/journal.pcbi.1007128].
Manubot then obtains reference metadata, exports the citations as Citation Style Language JSON Data Items, and renders the bibliographic information needed to generate the references section (2149).
This approach allows multiple authors to work on a piece of text without needing to make manual adjustments to the reference lists.
Due to the needs of this project, several new features were implemented in Manubot.
Because of the ever-evolving nature of the COVID-19 crisis, figures and statistics in the text quickly became outdated.
To address this concern, Manubot and GitHub’s CI features were used to create figures that integrated online data sources and to dynamically update information, such as the current number of active COVID-19 clinical trials (3), within the text of the manuscripts (Figure 15).
GitHub Actions runs a nightly workflow to update these external data and regenerate the statistics and figures for the manuscript.
The workflow uses the GitHub API to detect and save the latest commit of the external data sources that are GitHub repositories12.
It then downloads versioned data from that snapshot of the external repositories and runs bash and Python scripts to calculate the desired statistics and produce the summary figures using Matplotlib (2157).
The statistics are stored in JSON files that are accessed by Manubot to populate the values of placeholder template variables dynamically every time the manuscript is built.
For instance, the template variable {{ebm_trials_results}} in the manuscript is replaced by the actual number of clinical trials with results, 98.
The template variables also include versioned URLs to the dynamically updated figures.
The JSON files and figures are stored in the external-resources branch of the GitHub repository, providing versioned storage.
The GitHub Actions workflow automatically adds and commits the new JSON files and figures to the external-resources branch every time it runs, and Manubot uses the latest version of these resources when it builds the manuscript.
The GitHub Actions workflow file is available online13, as are the scripts14.
The Python package versions are also available15.
Figure 15:COVID-19 review GitHub repository organization and workflows.
Manubot uses CI to combine author-contributed content with automatically updated information from outside sources.
A nightly workflow updates figures and statistics derived from external resources.
Authors write text and add figures to the master branch (starred) via GitHub pull requests.
Manubot generates updated manuscript outputs for each new git commit, integrating the static text and figures with the dynamic statistics and figures and automatically-extracted citation information.
GitHub Pages hosts the latest HTML and PDF versions of the manuscript along with permanent links to prior versions.
Another issue identified was the need for standardized citation to clinical trials.
Other researchers identified the same need16.
Trials that are registered with clinicaltrials.gov receive a unique clinical trial identifier, or “NCT ID.”
Because clinical trials are registered long before results are published, referencing clinical trial identifiers was a priority.
Manubot uses the Zotero translation server17 to extract citation metadata for some types of citations.
However, Zotero did not support clinical trial identifiers and could not extract relevant metadata from their URLs.
In order to pull clinical trial metadata associated into Manubot, we added Zotero support for these identifiers.
To achieve this, we query clinicaltrials.gov to retrieve XML metadata associated with each identifier using JavaScript18.
This extension enables citing a trial as @clinicaltrials:NCT04280705 instead of the URL.
Then, when Manubot requests clinical trial metadata from the Zotero translation server, the response includes the trial sponsors, responsible investigators, title, and summary.
Manubot now supports directly citing hundreds of registered Compact Uniform Resource Identifiers19, beyond just the clinicaltrials identifier.
Because of the large number of citations used in this manuscript and the fast-moving nature of COVID-19 research, keeping track of retractions, corrections, and notices of concern also became a challenge.
We implemented a new Manubot plugin to support “smart citations” in the HTML build of manuscripts.
The plugin uses the scite (2158) service to display a badge below any citation with a DOI.
The badge contains a set of icons and numbers that indicate how many times that source has been mentioned, supported, or disputed and whether there have been any important editorial notices.
We were thus able to identify references that needed to be reevaluated by an expert.
This addition was invaluable given the nature of the project, where we were disseminating rapidly evolving information of great consequence from over 1,500 different sources.
The badges also allow readers to ascertain a rough approximation of the reliability of cited sources at a glance.
Because most collaborators were writing and editing text through the GitHub website rather than in a local text editor, we also needed to add spell-checking functionalities to Manubot.
We integrated an existing Pandoc20 spell-check extension with AppVeyor CI to automatically post spelling errors as comments in a GitHub pull request.
The comment reported both unique misspelled tokens and all locations where the token was detected.
Project maintainers managed a custom dictionary to allow over 1,500 scientific and technical terms that were not common English words.
Spell-checking also helped standardize the writing style across dozens of authors by detecting features such as British versus American English spellings.
The actual spell-checking was implemented using GNU Aspell21 and the Pandoc spellcheck filter22.
The filter enables checking only the manuscript text, ignoring URLs and formatting.
Manubot can render a manuscript in several formats that serve different purposes.
Prior to this project, Manubot could use Pandoc to convert the markdown-formatted manuscript to HTML, PDF, and DOCX formats.
We expanded this functionality to export individual sections of the manuscript as separate DOCX files while still rendering the complete manuscript in HTML and PDF formats.
This development was necessary because the manuscript grew so large that it needed to be split into seven separate papers for journal submission while still maintaining shared GitHub discussion across topics.
When exporting an individual section, Manubot customizes the manuscript title, authors, and author contributions to pertain to that specific section.
In addition, we expanded the export formats to include partial LaTeX support via Pandoc.
Pandoc converts the markdown content for an individual section to TeX and the Citation Style Language JSON, which contains reference metadata generated by Manubot, to BibTeX.
We customized a LaTeX template and reformatted the Manubot metadata, such as authors and their affiliations, for the LaTeX template.
The exported TeX file requires manual refinement but contains all manuscript content and most of the formatting.
Because LaTeX is required for manuscript submission in many fields, automating most of the process of converting markdown to a submission-friendly format expands Manubot’s potential user base.
Manubot users can write in the simple markdown format, render the manuscript in continuously-updated PDF or interactive HTML formats, and export the manuscript in DOCX or TeX and BibTeX for submission to traditional publishers, taking full advantage of Pandoc’s powerful document conversion capabilities and Manubot’s automation.
12.4 RESULTS
12.4.1 Recruitment and Manuscript Development
Figure 16:Project growth over time.
The number of authors, word count, and number of references have all grown dramatically from when the project began on March 20, 2020.
As of September 10, 2021, there were 52 authors (including consortia), 1676 references, and 138213 words.
The spike in word count during summer 2020 was caused by erroneous duplication and subsequent removal of a large appendix.
Coverage by Nature Toolbox(2159) and an associated tweet23 about the project on April 1, 2020 attracted the interest of the scientific community (Figure 16).
Because the GitHub issues and comment systems are similar to other common web commenting systems, authors learned these tools quickly.
The Gitter chat also presented a low barrier to entry.
The manuscript continued to grow throughout the first year and a half of the project in both word count and the number of references (Figure 16).
Though only a fraction of potential contributors contributed to the text included in the manuscripts (Figure 16), many contributors remained engaged over the long term (Figure 17).
Additionally, new contributors continued to join even into the second year of the project.
Figure 17:User contributions to the manuscript text over time.
The dot size indicates the number of words added or edited each month since March 2020.
The figure does not depict other types of author contributions such as literature summaries, pull request review, visualization, or software.
In order to make the project more accessible, we developed resources explaining how to use GitHub’s web interface to develop and edit text for Manubot assuming no prior experience working with version control.
These tutorials explained how to open an issue, open a pull request, and review a pull request24.
Additionally, the framework for evaluating literature was converted into issue templates to simplify the review of new articles.
Articles were classified as diagnostic, therapeutic, or other, with an associated template developed to guide the review of papers and preprints in each category.
A total of 285 new paper issues had been opened as of September 13, 2021.
The manuscripts produced by the consortium (excluding this one) will be submitted to mSystems as part of a special issue that provides support for continuous updates as more information becomes available.
One has been published and two are available as preprints.
This approach allows for a version of record to be maintained alongside the most recent version, which is always available through GitHub.
These manuscripts cover a wide range of topics including the fundamental biology of SARS-CoV-2 (pathogenesis (1) and evolution), biomedical advances in responding to the virus and COVID-19 (pharmaceuticals (3), nutraceuticals (2), vaccines, and diagnostic technologies), and biological and social factors influencing disease transmission and outcomes.
To date, 52 authors are associated with the consortium (Figure 16).
More formal recruitment efforts to integrate with existing projects providing support for undergraduate students during COVID-19 were also successful.
We incorporated summaries written by the students, post-docs, and faculty of the Immunology Institute at the Mount Sinai School of Medicine25(2143).
Additionally, two of the consortium authors were undergraduate students recruited through the American Physician Scientist Association’s Virtual Summer Research Program.
Thus, the consortium was successful in providing a venue for researchers across all career stages to continue investigating and publishing at a time when many biomedical researchers were unable to access their laboratory facilities.
12.4.2 Integrating Data
We integrated data into the manuscripts from several sources (Figure 15).
Worldwide cases and deaths were tracked by the COVID-19 Data Repository by the Center for Systems Science and Engineering at Johns Hopkins University26.
The clinical trials statistics and figure were generated based on data from the University of Oxford Evidence-Based Medicine Data Lab’s COVID-19 TrialsTracker (739).
Information about vaccine distribution was extracted from Our World In Data27(2160).
(Figure 14) integrates data from the CORD-19 dataset (2135).
Manubot’s bibliographic management capabilities were critical because the amount of relevant literature published far outstripped what we had anticipated at the beginning of the project.
As of September 10, 2021, there were 1676 references (Figure 16).
The scite plugin provided a way to visually inspect the reference list to identify possible references of concern.
This and the other new features required for the COVID-19 project are now included in Manubot’s rootstock, which is the template GitHub repository for creating a new manuscript.
Using CI, Manubot now checks that the manuscript was built correctly, runs spell-checking, and cross-references the manuscripts cited in this review.
In addition, Manubot rootstock now supports citing clinical trial identifiers such as clinicaltrials:NCT04292899(809).
12.5 DISCUSSION
The current project was based in the GitHub repository greenelab/covid19-review using Manubot (2149) to continuously generate the manuscript.
The Manubot framework facilitated a massive collaborative review on an urgent topic.
We demonstrated the utility of Manubot to a project where many contributors lacked expertise or even experience working with version control.
This effort has produced not only seven literature reviews on topics relevant to the COVID-19 pandemic, but has also generated cyberinfrastructure for training novice users in GitHub.
We also extended the functionalities of Manubot to provide more of the benefits of What You See Is What You Get platforms such as Google Docs (Table 6).
Open publishing thus allowed us to harness the domain expertise of a large group of non-technical users to respond to the flood of COVID-19 publications.
Several existing and new features in Manubot aid in responding to the challenges posed by the infodemic.
Manuscripts are written in markdown and can be rendered in several formats providing different advantages to users.
For example, beyond building just a PDF, Manubot also renders the manuscript in HTML, DOCX, and now, LaTeX (in a more limited capacity).
The interactive HTML manuscript format offers several advantages over a static PDF to harmonize available resources and address specific problems related to COVID-19.
The integration of scite into the HTML build makes references more manageable by visually indicating whether their results are contested or whether they have been corrected or retracted.
Cross-referencing different pieces of the manuscript, such as cited preprints with reviews stored in an appendix, is another interactive option presented by HTML.
The DOCX format was preferred by most non-technical users for reviewing the final version of the manuscript and was useful for creating submissions to a biological journal.
Additionally, because of the heavy emphasis on Word processing in biology, Manubot’s ability to generate DOCX outputs was expanded to allow users to generate DOCX files containing only a section of the manuscript.
In our case, where the full project is nearly 150,000 words, this allows individual pieces to be shared more easily.
Finally, the preliminary addition of LaTeX output is useful for researchers from computational fields who submit papers in TeX format and removes the step of reformatting markdown prior to submission.
Table 6: Manubot extensions for the COVID-19 review.
Type
Description
CI
Regularly download external data sources, generate new figures and statistics, and read them when Manubot builds the latest manuscript
CI
Post spell-checking reports as pull request comments
Cite any Compact Uniform Resource Identifier, such as clinicaltrials or ncbigene
Citations
scite badges to track retractions, corrections, and notices of concern
Outputs
Improved support for Pandoc’s LaTeX output
Outputs
Build complete manuscript alongside individual sections as standalone documents
The COVID-19 Review Consortium provided a platform for researchers to engage in scientific investigation early in the pandemic when many biological scientists were unable to access their research spaces.
In turn, by seeking to adapt Manubot to allow for broader participation, we made a number of improvements that are expected to increase its appeal to researchers from all backgrounds.
Manubot provided a way for contributors from a variety of backgrounds, including early-career researchers, to join a massive collaborative project while demonstrating their individual contributions to the larger work and gaining experience with version control.
The licensing and infrastructure also provide the basis for individuals to adapt from this project to create their own snapshots of the COVID-19 literature that derive from, but are not wholly identical to, the primary versions of these reviews.
This project suggests that massive online open publishing efforts can indeed advance scholarship through inclusion (2150), including during the extreme challenges presented by the COVID-19 pandemic.
Some challenges did arise in efforts to include an academically diverse set of authors.
The barriers to entry posed by git and GitHub likely still reduced participation from individuals who might have otherwise been interested.
Using pull requests as a tool for writing text is also unfamiliar to many or most scientists, and the review process can be slow, which might cause interested contributors to lose interest.
Additionally, the pull request model may limit people from providing general feedback on the manuscript or a section of the manuscript, unless there is an open pull request.
As a result, some feedback came through email or comments on the DOCX outputs that were then translated into issues or pull requests by the project managers.
Given that our approach hinged on these version control tools, it is likely that our group of contributors was biased towards those who were interested in or experienced with computational tools.
The trajectory of the pandemic itself also likely influenced participation: engagement waned over the course of the pandemic as labs opened back up and researchers were able to return to their work, and we recruited very few senior clinicians to the project, which is unsurprising given the load on medical professionals during this time.
Engagement that waxes and wanes is, however, typical when writing massively open online papers (2150).
Adding features such as spell-check did improve usability, and additional features such as automatically checking the formatting of citations could further improve the usability of this tool.
In the future, a formal study of participation could allow for quantification of these biases and improved efforts to foster inclusion.
Additional limitations are challenges associated with massively open online papers in general.
With such a large amount of text, it is not possible to keep all sections of the manuscript up to date at all times.
Readers are not able to distinguish when each section was updated.
Even GitHub’s blame functionality does not distinguish minor changes from substantive updates to the text.
While much of the data and statistics update automatically, the text itself required updating by human experts.
This asynchronicity could potentially introduce incompatibility between the figures and the surrounding text.
Similarly, in line with the collaboration-related challenges of the project, some authors returned to update their text, while others did not.
As a result, the lead authors of each paper often spent several weeks prior to journal submission updating the text to reflect new developments in each area.
In the future, it may be possible to streamline this process through integration with a tool such as CoronaCentral (2136) to automatically identify relevant, high-impact papers that need to be included, although expertise would still be required to incorporate them.
Another challenge involves tracking preprints as they are reviewed or critiqued, revised, and potentially published.
While updating the content of the manuscript would likely fall to human contributors, automatic detection of published versions of preprints (2161) could be integrated in the future.
These challenges are exacerbated by the scale of the infodemic, but developing solutions would benefit future projects tracking more typical trends in publication.
Similarly, outputting machine readable summaries of key information in the COVID-19 review manuscripts could reduce their contribution to the infodemic.
As it stands, the integration of Compact Uniform Resource Identifier does make a step in this direction.
Formal identifiers could be used to extract relationships among clinical trials, genes, publications, and other entities.
Thus, the experience of using Manubot for a massive project has laid the foundation for future additions to enhance user experience and inclusivity.
12.6 CONCLUSION
With the worldwide scientific community uniting during 2020 and 2021 to investigate COVID-19 from a wide range of perspectives, findings from many disciplines are relevant on a rapid timescale to a broad scientific audience.
As many other efforts have described, the publishing rate of formal manuscripts and preprints about COVID-19 has been unprecedented (2133), and efforts to review the body of COVID-19 literature are faced with an ever-expanding corpus to evaluate.
In the case of the seven manuscripts produced by the COVID-19 Review Consortium, Manubot allows for continuous updating of the manuscripts as the pandemic enters its second year and the landscape shifts with the emergence of promising therapeutics and vaccines (3).
These manuscripts pull data from external sources and update information and visualizations daily using CI.
By off-loading some updates to computational pipelines, domain experts can focus on the broader implications of new information as it emerges.
Centralizing, summarizing, and critiquing data and literature broadly relevant to COVID-19 can expedite the interdisciplinary scientific process that is currently happening at an advanced pace.
As of September 13, 2021, 2886 commits have been made to the manuscript across 575 merged pull requests.
The efforts of the COVID-19 Review Consortium illustrate the value of including open source tools, including those focused on open publishing, in these efforts.
By facilitating the versioning of text, such platforms also allow for documentation of the evolution of thought in an evolving area and formal analysis of a collaborative project.
This application of version control holds the potential to improve scientific publishing in a range of disciplines, including those outside of traditional computational fields.
While Manubot is a technologically complex tool, this project demonstrates that it can be applied to a variety of projects.
Future work can address remaining limitations and continue to advance Manubot as an inclusive tool for open publishing projects.
13 Additional Items
13.1 Competing Interests
Author
Competing Interests
Last Reviewed
Halie M. Rando
None
2021-01-20
Casey S. Greene
None
2021-01-20
Michael P. Robson
None
2020-11-12
Simina M. Boca
Currently an employee at AstraZeneca, Gaithersburg, MD, USA, may own stock or stock options.
2021-07-01
Nils Wellhausen
None
2020-11-03
Ronan Lordan
None
2020-11-03
Christian Brueffer
Shareholder of SAGA Diagnostics AB.
2023-03-06
Sandipan Ray
None
2020-11-11
Lucy D'Agostino McGowan
Received consulting fees from Acelity and Sanofi in the past five years
2020-11-10
Anthony Gitter
Inventor of patent US-11410440-B2 assigned to the Wisconsin Alumni Research Foundation related to classifying activated T cells.
2023-01-11
Anna Ada Dattoli
None
2020-03-26
Ryan Velazquez
None
2020-11-10
John P. Barton
None
2020-11-11
Jeffrey M. Field
None
2020-11-12
Bharath Ramsundar
None
2020-11-11
Adam L. MacLean
None
2021-02-23
Alexandra J. Lee
None
2020-11-09
Immunology Institute of the Icahn School of Medicine
None
2020-04-07
Fengling Hu
None
2020-04-08
Nafisa M. Jadavji
None
2020-11-11
Elizabeth Sell
None
2020-11-11
Vincent Rubinetti
None
2021-04-29
Jinhui Wang
None
2021-01-21
Diane N. Rafizadeh
None
2020-11-11
Ashwin N. Skelly
None
2020-11-11
Marouen Ben Guebila
None
2021-08-02
Likhitha Kolla
None
2020-11-16
David Manheim
None
2022-03-15
Soumita Ghosh
None
2020-11-09
James Brian Byrd
Funded by FastGrants to conduct a COVID-19-related clinical trial
2020-11-12
YoSon Park
YoSon Park is affiliated with Pfizer Worldwide Research. The author has no financial interests to declare and contributed as an author prior to joining Pfizer, and the work was not part of a Pfizer collaboration nor was it funded by Pfizer.
2020-01-22
Vikas Bansal
None
2021-01-25
Stephen Capone
None
2020-11-11
John J. Dziak
None
2020-11-11
Yuchen Sun
None
2020-11-11
Yanjun Qi
None
2020-07-09
Lamonica Shinholster
None
2020-11-11
Temitayo Lukan
None
2020-11-10
Sergey Knyazev
None
2020-11-11
Dimitri Perrin
None
2020-11-11
Serghei Mangul
None
2020-11-11
Shikta Das
None
2020-08-13
Gregory L Szeto
None
2020-11-16
Tiago Lubiana
None
2020-11-11
David Mai
None
2021-01-08
COVID-19 Review Consortium
None
2021-01-16
Rishi Raj Goel
None
2021-01-20
Joel D Boerckel
None
2021-03-26
Amruta Naik
None
2021-04-05
Yusha Sun
None
2021-04-10
Daniel S. Himmelstein
None
2021-04-30
Jeremy P. Kamil
None
2022-10-11
Jesse G. Meyer
None
2022-01-06
Ariel I. Mundo
None
2021-12-19
13.2 Author Contributions
Author
Contributions
Halie M. Rando
D, E, Project Administration, Software, Visualization, Writing - Original Draft, Writing - Review & Editing
Writing - Original Draft, Writing - Review & Editing
Marouen Ben Guebila
Writing - Original Draft, Writing - Review & Editing
Likhitha Kolla
Writing - Original Draft, Writing - Review & Editing
David Manheim
Writing - Original Draft, Writing - Review & Editing
Soumita Ghosh
Writing - Original Draft
James Brian Byrd
Writing - Original Draft, Writing - Review & Editing
YoSon Park
Writing - Original Draft, Writing - Review & Editing
Vikas Bansal
Writing - Original Draft, Writing - Review & Editing
Stephen Capone
Writing - Original Draft, Writing - Review & Editing
John J. Dziak
Writing - Original Draft, Writing - Review & Editing
Yuchen Sun
Visualization
Yanjun Qi
Visualization
Lamonica Shinholster
Writing - Original Draft
Temitayo Lukan
Investigation, Writing - Original Draft
Sergey Knyazev
Writing - Original Draft, Writing - Review & Editing
Dimitri Perrin
Writing - Original Draft, Writing - Review & Editing
Serghei Mangul
Writing - Review & Editing
Shikta Das
Writing - Review & Editing
Gregory L Szeto
Writing - Review & Editing
Tiago Lubiana
Writing - Review & Editing
David Mai
Writing - Original Draft, Writing - Review & Editing
COVID-19 Review Consortium
Project Administration
Rishi Raj Goel
Writing - Original Draft, Writing - Review & Editing
Joel D Boerckel
Writing - Review & Editing
Amruta Naik
Writing - Original Draft, Writing - Review & Editing
Yusha Sun
Writing - Review & Editing
Daniel S. Himmelstein
Software
Jeremy P. Kamil
Writing - Review & Editing
Jesse G. Meyer
Writing - Original Draft, Writing - Review & Editing
Ariel I. Mundo
Writing - Original Draft, Writing - Review & Editing
13.3 Acknowledgements
We thank Nick DeVito for assistance with the Evidence-Based Medicine Data Lab COVID-19 TrialsTracker data.
We thank Yael Evelyn Marshall who contributed writing (original draft) as well as reviewing and editing of pieces of the text but who did not formally approve the manuscript, as well as Ronnie Russell, who contributed text to and helped develop the structure of the manuscript early in the writing process and Matthias Fax who helped with writing and editing text related to diagnostics.
We are also very grateful to James Fraser for suggestions about successes and limitations in the area of computational screening for drug repurposing.
We are grateful to the following contributors for reviewing pieces of the text: Nadia Danilova, James Eberwine and Ipsita Krishnan.
13.4 References
1.
Rando HM, MacLean AL, Lee AJ, Lordan R, Ray S, Bansal V, Skelly AN, Sell E, Dziak JJ, Shinholster L, D'Agostino McGowan L, Ben Guebila M, Wellhausen N, Knyazev S, Boca SM, Capone S, Qi Y, Park Y, Mai D, Sun Y, Boerckel JD, Brueffer C, Byrd JB, Kamil JP, Wang J, Velazquez R, Szeto GL, Barton JP, Goel RR, Mangul S, Lubiana T, COVID-19 Review Consortium Vikas Bansal, John P. Barton, Simina M. Boca, Joel D. Boerckel, Christian Brueffer, James Brian Byrd, Stephen Capone, Shikta Das, Anna Ada Dattoli, John J. Dziak, Jeffrey M. Field, Soumita Ghosh, Anthony Gitter, Rishi Raj Goel, Casey S. Greene, Marouen Ben Guebila, Daniel S. Himmelstein, Fengling Hu, Nafisa M. Jadavji, Jeremy P. Kamil, Sergey Knyazev, Likhitha Kolla, Alexandra J. Lee, Ronan Lordan, Tiago Lubiana, Temitayo Lukan, Adam L. MacLean, David Mai, Serghei Mangul, David Manheim, Lucy D’Agostino McGowan, Amruta Naik, YoSon Park, Dimitri Perrin, Yanjun Qi, Diane N. Rafizadeh, Bharath Ramsundar, Halie M. Rando, Sandipan Ray, Michael P. Robson, Vincent Rubinetti, Elizabeth Sell, Lamonica Shinholster, Ashwin N. Skelly, Yuchen Sun, Yusha Sun, Gregory L. Szeto, Ryan Velazquez, Jinhui Wang, Nils Wellhausen, Gitter A, Greene CS. 2021. Pathogenesis, Symptomatology, and Transmission of SARS-CoV-2 through Analysis of Viral Genomics and Structure. mSystems 6:e0009521.
Rando HM, Wellhausen N, Ghosh S, Lee AJ, Dattoli AA, Hu F, Byrd JB, Rafizadeh DN, Lordan R, Qi Y, Sun Y, Brueffer C, Field JM, Ben Guebila M, Jadavji NM, Skelly AN, Ramsundar B, Wang J, Goel RR, Park Y, COVID-19 Review Consortium Vikas Bansal, John P. Barton, Simina M. Boca, Joel D. Boerckel, Christian Brueffer, James Brian Byrd, Stephen Capone, Shikta Das, Anna Ada Dattoli, John J. Dziak, Jeffrey M. Field, Soumita Ghosh, Anthony Gitter, Rishi Raj Goel, Casey S. Greene, Marouen Ben Guebila, Daniel S. Himmelstein, Fengling Hu, Nafisa M. Jadavji, Jeremy P. Kamil, Sergey Knyazev, Likhitha Kolla, Alexandra J. Lee, Ronan Lordan, Tiago Lubiana, Temitayo Lukan, Adam L. MacLean, David Mai, Serghei Mangul, David Manheim, Lucy D’Agostino McGowan, Amruta Naik, YoSon Park, Dimitri Perrin, Yanjun Qi, Diane N. Rafizadeh, Bharath Ramsundar, Halie M. Rando, Sandipan Ray, Michael P. Robson, Vincent Rubinetti, Elizabeth Sell, Lamonica Shinholster, Ashwin N. Skelly, Yuchen Sun, Yusha Sun, Gregory L. Szeto, Ryan Velazquez, Jinhui Wang, Nils Wellhausen, Boca SM, Gitter A, Greene CS. 2021. Identification and Development of Therapeutics for COVID-19. mSystems 6:e0023321.
4.
Rando HM, Boca SM, D'Agostino McGowan L, Himmelstein DS, Robson MP, Rubinetti V, Velazquez R, Greene CS, Gitter A. 2021. An Open-Publishing Response to the COVID-19 Infodemic. Proceedings of the Workshop on Digital Infrastructures for Scholarly Content Objects (DISCO 2021) 2976:29–38.
Tan W, Zhao X, Ma X, Wang W, Niu P, Xu W, F. Gao G, Wu G, MHC Key Laboratory of Biosafety, National Institute for Viral Disease Control and Prevention, China CDC, Beijing, China, Center for Biosafety Mega-Science, Chinese Academy of Sciences, Beijing, China. 2020. A Novel Coronavirus Genome Identified in a Cluster of Pneumonia Cases — Wuhan, China 2019−2020. China CDC Weekly 2:61–62.
Johnson BA, Xie X, Kalveram B, Lokugamage KG, Muruato A, Zou J, Zhang X, Juelich T, Smith JK, Zhang L, Bopp N, Schindewolf C, Vu M, Vanderheiden A, Swetnam D, Plante JA, Aguilar P, Plante KS, Lee B, Weaver SC, Suthar MS, Routh AL, Ren P, Ku Z, An Z, Debbink K, Shi PY, Freiberg AN, Menachery VD. 2020. Furin Cleavage Site Is Key to SARS-CoV-2 Pathogenesis. Cold Spring Harbor Laboratory.
38.
Johnson BA, Xie X, Bailey AL, Kalveram B, Lokugamage KG, Muruato A, Zou J, Zhang X, Juelich T, Smith JK, Zhang L, Bopp N, Schindewolf C, Vu M, Vanderheiden A, Winkler ES, Swetnam D, Plante JA, Aguilar P, Plante KS, Popov V, Lee B, Weaver SC, Suthar MS, Routh AL, Ren P, Ku Z, An Z, Debbink K, Diamond MS, Shi P-Y, Freiberg AN, Menachery VD. 2021. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 591:293–299.
2020. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pacific Journal of Allergy and Immunology https://doi.org/10.12932/ap-200220-0772.
Wang W, Xu Y, Gao R, Lu R, Han K, Wu G, Tan W. 2020. Detection of SARS-CoV-2 in Different Types of Clinical Specimens. JAMA https://doi.org/10.1001/jama.2020.3786.
72.
Young BE, Ong SWX, Kalimuddin S, Low JG, Tan SY, Loh J, Ng O-T, Marimuthu K, Ang LW, Mak TM, Lau SK, Anderson DE, Chan KS, Tan TY, Ng TY, Cui L, Said Z, Kurupatham L, Chen MI-C, Chan M, Vasoo S, Wang L-F, Tan BH, Lin RTP, Lee VJM, Leo Y-S, Lye DC, for the Singapore 2019 Novel Coronavirus Outbreak Research Team. 2020. Epidemiologic Features and Clinical Course of Patients Infected With SARS-CoV-2 in Singapore. JAMA 323:1488.
Agarwal A, Rochwerg B, Lamontagne F, Siemieniuk RA, Agoritsas T, Askie L, Lytvyn L, Leo Y-S, Macdonald H, Zeng L, Amin W, da Silva ARA, Aryal D, Barragan FAJ, Bausch FJ, Burhan E, Calfee CS, Cecconi M, Chacko B, Chanda D, Dat VQ, De Sutter A, Du B, Freedman S, Geduld H, Gee P, Gotte M, Harley N, Hashmi M, Hunt B, Jehan F, Kabra SK, Kanda S, Kim Y-J, Kissoon N, Krishna S, Kuppalli K, Kwizera A, Castro-Rial ML, Lisboa T, Lodha R, Mahaka I, Manai H, Mendelson M, Battista Migliori G, Mino G, Nsutebu E, Preller J, Pshenichnaya N, Qadir N, Relan P, Sabzwari S, Sarin R, Shankar-Hari M, Sharland M, Shen Y, Ranganathan SS, Souza JP, Stegemann M, Swanstrom R, Ugarte S, Uyeki T, Venkatapuram S, Vuyiseka D, Wijewickrama A, Tran L, Zeraatkar D, Bartoszko JJ, Ge L, Brignardello-Petersen R, Owen A, Guyatt G, Diaz J, Kawano-Dourado L, Jacobs M, Vandvik PO. 2020. A living WHO guideline on drugs for covid-19. BMJ m3379.
82.
Guan W, Ni Z, Hu Y, Liang W, Ou C, He J, Liu L, Shan H, Lei C, Hui DSC, Du B, Li L, Zeng G, Yuen K-Y, Chen R, Tang C, Wang T, Chen P, Xiang J, Li S, Wang J, Liang Z, Peng Y, Wei L, Liu Y, Hu Y, Peng P, Wang J, Liu J, Chen Z, Li G, Zheng Z, Qiu S, Luo J, Ye C, Zhu S, Zhong N. 2020. Clinical Characteristics of Coronavirus Disease 2019 in China. New England Journal of Medicine 382:1708–1720.
Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW, Barnaby DP, Becker LB, Chelico JD, Cohen SL, Cookingham J, Coppa K, Diefenbach MA, Dominello AJ, Duer-Hefele J, Falzon L, Gitlin J, Hajizadeh N, Harvin TG, Hirschwerk DA, Kim EJ, Kozel ZM, Marrast LM, Mogavero JN, Osorio GA, Qiu M, Zanos TP, and the Northwell COVID-19 Research Consortium. 2020. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 323:2052.
85.
Goyal P, Choi JJ, Pinheiro LC, Schenck EJ, Chen R, Jabri A, Satlin MJ, Campion TR, Nahid M, Ringel JB, Hoffman KL, Alshak MN, Li HA, Wehmeyer GT, Rajan M, Reshetnyak E, Hupert N, Horn EM, Martinez FJ, Gulick RM, Safford MM. 2020. Clinical Characteristics of Covid-19 in New York City. New England Journal of Medicine 382:2372–2374.
86.
Burke RM, Killerby ME, Newton S, Ashworth CE, Berns AL, Brennan S, Bressler JM, Bye E, Crawford R, Harduar Morano L, Lewis NM, Markus TM, Read JS, Rissman T, Taylor J, Tate JE, Midgley CM, Balachandran N, Dahl RM, Dott M, Gilani Z, Grober A, Leung J, O’Hegarty M, Person J, Ricaldi JN, Roth NM, Sejvar JJ, Shimabukuro T, Tran CH, Watson JT, Whitham H, Chiou H, Clogher P, Duca LM, Dratch A, Feldpausch A, Fill M-M, Ghinai I, Holshue M, Scott S, Westergaard R, Case Investigation Form Working Group, Case Investigation Form Working Group. 2020. Symptom Profiles of a Convenience Sample of Patients with COVID-19 — United States, January–April 2020. MMWR Morbidity and Mortality Weekly Report 69:904–908.
87.
Allen WE, Altae-Tran H, Briggs J, Jin X, McGee G, Shi A, Raghavan R, Kamariza M, Nova N, Pereta A, Danford C, Kamel A, Gothe P, Milam E, Aurambault J, Primke T, Li W, Inkenbrandt J, Huynh T, Chen E, Lee C, Croatto M, Bentley H, Lu W, Murray R, Travassos M, Coull BA, Openshaw J, Greene CS, Shalem O, King G, Probasco R, Cheng DR, Silbermann B, Zhang F, Lin X. 2020. Population-scale longitudinal mapping of COVID-19 symptoms, behaviour and testing. Nat Hum Behav 4:972–982.
88.
Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, Bikdeli B, Ahluwalia N, Ausiello JC, Wan EY, Freedberg DE, Kirtane AJ, Parikh SA, Maurer MS, Nordvig AS, Accili D, Bathon JM, Mohan S, Bauer KA, Leon MB, Krumholz HM, Uriel N, Mehra MR, Elkind MSV, Stone GW, Schwartz A, Ho DD, Bilezikian JP, Landry DW. 2020. Extrapulmonary manifestations of COVID-19. Nature Medicine 26:1017–1032.
89.
Hirsch JS, Ng JH, Ross DW, Sharma P, Shah HH, Barnett RL, Hazzan AD, Fishbane S, Jhaveri KD, Abate M, Andrade HP, Barnett RL, Bellucci A, Bhaskaran MC, Corona AG, Chang BF, Finger M, Fishbane S, Gitman M, Halinski C, Hasan S, Hazzan AD, Hirsch JS, Hong S, Jhaveri KD, Khanin Y, Kuan A, Madireddy V, Malieckal D, Muzib A, Nair G, Nair VV, Ng JH, Parikh R, Ross DW, Sakhiya V, Sachdeva M, Schwarz R, Shah HH, Sharma P, Singhal PC, Uppal NN, Wanchoo R, Bessy Suyin Flores Chang, Ng JHwei. 2020. Acute kidney injury in patients hospitalized with COVID-19. Kidney International 98:209–218.
Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, Kneen R, Defres S, Sejvar J, Solomon T. 2020. Neurological associations of COVID-19. The Lancet Neurology 19:767–783.
Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, Laue M, Schneider J, Brünink S, Greuel S, Lehmann M, Hassan O, Aschman T, Schumann E, Chua RL, Conrad C, Eils R, Stenzel W, Windgassen M, Rößler L, Goebel H-H, Gelderblom HR, Martin H, Nitsche A, Schulz-Schaeffer WJ, Hakroush S, Winkler MS, Tampe B, Scheibe F, Körtvélyessy P, Reinhold D, Siegmund B, Kühl AA, Elezkurtaj S, Horst D, Oesterhelweg L, Tsokos M, Ingold-Heppner B, Stadelmann C, Drosten C, Corman VM, Radbruch H, Heppner FL. 2020. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nature Neuroscience 24:168–175.
97.
Thakur KT, Miller EH, Glendinning MD, Al-Dalahmah O, Banu MA, Boehme AK, Boubour AL, Bruce SS, Chong AM, Claassen J, Faust PL, Hargus G, Hickman RA, Jambawalikar S, Khandji AG, Kim CY, Klein RS, Lignelli-Dipple A, Lin C-C, Liu Y, Miller ML, Moonis G, Nordvig AS, Overdevest JB, Prust ML, Przedborski S, Roth WH, Soung A, Tanji K, Teich AF, Agalliu D, Uhlemann A-C, Goldman JE, Canoll P. 2021. COVID-19 neuropathology at Columbia University Irving Medical Center/New York Presbyterian Hospital. Brain 144:2696–2708.
98.
Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, De Leacy RA, Shigematsu T, Ladner TR, Yaeger KA, Skliut M, Weinberger J, Dangayach NS, Bederson JB, Tuhrim S, Fifi JT. 2020. Large-Vessel Stroke as a Presenting Feature of Covid-19 in the Young. New England Journal of Medicine 382:e60.
Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, Chen H, Ding X, Zhao H, Zhang H, Wang C, Zhao J, Sun X, Tian R, Wu W, Wu D, Ma J, Chen Y, Zhang D, Xie J, Yan X, Zhou X, Liu Z, Wang J, Du B, Qin Y, Gao P, Qin X, Xu Y, Zhang W, Li T, Zhang F, Zhao Y, Li Y, Zhang S. 2020. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. New England Journal of Medicine 382:e38.
Huang C, Huang L, Wang Y, Li X, Ren L, Gu X, Kang L, Guo L, Liu M, Zhou X, Luo J, Huang Z, Tu S, Zhao Y, Chen L, Xu D, Li Y, Li C, Peng L, Li Y, Xie W, Cui D, Shang L, Fan G, Xu J, Wang G, Wang Y, Zhong J, Wang C, Wang J, Zhang D, Cao B. 2021. 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. The Lancet 397:220–232.
Deer RR, Rock MA, Vasilevsky N, Carmody L, Rando H, Anzalone AJ, Callahan TJ, Bramante CT, Chute CG, Greene CS, Gagnier J, Chu H, Koraishy FM, Liang C, Liu F, Madlock-Brown CR, Mazzotti DR, McNair DS, Parker AM, Coleman BD, Davis HE, Perry MA, Reese JT, Saltz J, Solomonides AE, Sule AA, Stein GS, Köhler S, Monteith TS, Madhira V, Kimble WD, Kavuluru R, Hillegass WB, Chan LE, Byrd JB, Boudreau EA, Liu H, McMurry JA, Pfaff E, Matentzoglu N, Relevo R, Moffitt RA, Schuff RA, Solway J, Spratt H, Bergquist T, Bennett TD, Basson MD, Topaloglu U, Wang L, Haendel MA, Robinson PN. 2021. Characterizing Long COVID: Deep Phenotype of a Complex Condition. Cold Spring Harbor Laboratory.
115.
Fontanet A, Grant R, Tondeur L, Madec Y, Grzelak L, Cailleau I, Ungeheuer M-N, Renaudat C, Pellerin SF, Kuhmel L, Staropoli I, Anna F, Charneau P, Demeret C, Bruel T, Schwartz O, Hoen B. 2020. SARS-CoV-2 infection in primary schools in northern France: A retrospective cohort study in an area of high transmission. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.06.25.20140178.
116.
Lu X, Zhang L, Du H, Zhang J, Li YY, Qu J, Zhang W, Wang Y, Bao S, Li Y, Wu C, Liu H, Liu D, Shao J, Peng X, Yang Y, Liu Z, Xiang Y, Zhang F, Silva RM, Pinkerton KE, Shen K, Xiao H, Xu S, Wong GWK. 2020. SARS-CoV-2 Infection in Children. New England Journal of Medicine 382:1663–1665.
Diorio C, Henrickson SE, Vella LA, McNerney KO, Chase J, Burudpakdee C, Lee JH, Jasen C, Balamuth F, Barrett DM, Banwell BL, Bernt KM, Blatz AM, Chiotos K, Fisher BT, Fitzgerald JC, Gerber JS, Gollomp K, Gray C, Grupp SA, Harris RM, Kilbaugh TJ, John ARO, Lambert M, Liebling EJ, Paessler ME, Petrosa W, Phillips C, Reilly AF, Romberg ND, Seif A, Sesok-Pizzini DA, Sullivan KE, Vardaro J, Behrens EM, Teachey DT, Bassiri H. 2020. Multisystem inflammatory syndrome in children and COVID-19 are distinct presentations of SARS–CoV-2. Journal of Clinical Investigation 130:5967–5975.
131.
Consiglio CR, Cotugno N, Sardh F, Pou C, Amodio D, Rodriguez L, Tan Z, Zicari S, Ruggiero A, Pascucci GR, Santilli V, Campbell T, Bryceson Y, Eriksson D, Wang J, Marchesi A, Lakshmikanth T, Campana A, Villani A, Rossi P, Landegren N, Palma P, Brodin P. 2020. The Immunology of Multisystem Inflammatory Syndrome in Children with COVID-19. Cell 183:968–981.e7.
132.
Belhadjer Z, Méot M, Bajolle F, Khraiche D, Legendre A, Abakka S, Auriau J, Grimaud M, Oualha M, Beghetti M, Wacker J, Ovaert C, Hascoet S, Selegny M, Malekzadeh-Milani S, Maltret A, Bosser G, Giroux N, Bonnemains L, Bordet J, Di Filippo S, Mauran P, Falcon-Eicher S, Thambo J-B, Lefort B, Moceri P, Houyel L, Renolleau S, Bonnet D. 2020. Acute Heart Failure in Multisystem Inflammatory Syndrome in Children in the Context of Global SARS-CoV-2 Pandemic. Circulation 142:429–436.
Feldstein LR, Tenforde MW, Friedman KG, Newhams M, Rose EB, Dapul H, Soma VL, Maddux AB, Mourani PM, Bowens C, Maamari M, Hall MW, Riggs BJ, Giuliano JS Jr, Singh AR, Li S, Kong M, Schuster JE, McLaughlin GE, Schwartz SP, Walker TC, Loftis LL, Hobbs CV, Halasa NB, Doymaz S, Babbitt CJ, Hume JR, Gertz SJ, Irby K, Clouser KN, Cvijanovich NZ, Bradford TT, Smith LS, Heidemann SM, Zackai SP, Wellnitz K, Nofziger RA, Horwitz SM, Carroll RW, Rowan CM, Tarquinio KM, Mack EH, Fitzgerald JC, Coates BM, Jackson AM, Young CC, Son MBF, Patel MM, Newburger JW, Randolph AG. 2021. Characteristics and Outcomes of US Children and Adolescents With Multisystem Inflammatory Syndrome in Children (MIS-C) Compared With Severe Acute COVID-19. JAMA 325:1074.
Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. 2012. Into the Eye of the Cytokine Storm. Microbiology and Molecular Biology Reviews 76:16–32.
Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, Kochanek M, Böll B, von Bergwelt-Baildon MS. 2018. Cytokine release syndrome. Journal for ImmunoTherapy of Cancer 6:56.
Wang P-H, Cheng Y. 2020. Increasing Host Cellular Receptor—Angiotensin-Converting Enzyme 2 (ACE2) Expression by Coronavirus may Facilitate 2019-nCoV Infection. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.24.963348.
Emanuel W, Kirstin M, Vedran F, Asija D, Theresa GL, Roberto A, Filippos K, David K, Salah A, Christopher B, Anja R, Ivano L, Andranik I, Tommaso M, Simone DG, Patrick PJ, Alexander MM, Daniela N, Matthias S, Altuna A, Nikolaus R, Christian D, Markus L. 2020. Bulk and single-cell gene expression profiling of SARS-CoV-2 infected human cell lines identifies molecular targets for therapeutic intervention. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.05.05.079194.
176.
Harcourt J, Tamin A, Lu X, Kamili S, Sakthivel SKumar, Murray J, Queen K, Tao Y, Paden CR, Zhang J, Li Y, Uehara A, Wang H, Goldsmith C, Bullock HA, Wang L, Whitaker B, Lynch B, Gautam R, Schindewolf C, Lokugamage KG, Scharton D, Plante JA, Mirchandani D, Widen SG, Narayanan K, Makino S, Ksiazek TG, Plante KS, Weaver SC, Lindstrom S, Tong S, Menachery VD, Thornburg NJ. 2020. Isolation and characterization of SARS-CoV-2 from the first US COVID-19 patient. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.02.972935.
177.
Aschenbrenner AC, Mouktaroudi M, Krämer B, Oestreich M, Antonakos N, Nuesch-Germano M, Gkizeli K, Bonaguro L, Reusch N, Baßler K, Saridaki M, Knoll R, Pecht T, Kapellos TS, Doulou S, Kröger C, Herbert M, Holsten L, Horne A, Gemünd ID, Rovina N, Agrawal S, Dahm K, van Uelft M, Drews A, Lenkeit L, Bruse N, Gerretsen J, Gierlich J, Becker M, Händler K, Kraut M, Theis H, Mengiste S, De Domenico E, Schulte-Schrepping J, Seep L, Raabe J, Hoffmeister C, ToVinh M, Keitel V, Rieke G, Talevi V, Skowasch D, Aziz NA, Pickkers P, van de Veerdonk FL, Netea MG, Schultze JL, Kox M, Breteler MMB, Nattermann J, Koutsoukou A, Giamarellos-Bourboulis EJ, Ulas T. 2021. Disease severity-specific neutrophil signatures in blood transcriptomes stratify COVID-19 patients. Genome Med 13.
178.
Bernardes JP, Mishra N, Tran F, Bahmer T, Best L, Blase JI, Bordoni D, Franzenburg J, Geisen U, Josephs-Spaulding J, Köhler P, Künstner A, Rosati E, Aschenbrenner AC, Bacher P, Baran N, Boysen T, Brandt B, Bruse N, Dörr J, Dräger A, Elke G, Ellinghaus D, Fischer J, Forster M, Franke A, Franzenburg S, Frey N, Friedrichs A, Fuß J, Glück A, Hamm J, Hinrichsen F, Hoeppner MP, Imm S, Junker R, Kaiser S, Kan YH, Knoll R, Lange C, Laue G, Lier C, Lindner M, Marinos G, Markewitz R, Nattermann J, Noth R, Pickkers P, Rabe KF, Renz A, Röcken C, Rupp J, Schaffarzyk A, Scheffold A, Schulte-Schrepping J, Schunk D, Skowasch D, Ulas T, Wandinger K-P, Wittig M, Zimmermann J, Busch H, Hoyer BF, Kaleta C, Heyckendorf J, Kox M, Rybniker J, Schreiber S, Schultze JL, Rosenstiel P, Banovich NE, Desai T, Eickelberg O, Haniffa M, Horvath P, Kropski JA, Lafyatis R, Lundeberg J, Meyer K, Nawijn MC, Nikolic M, Ordovas Montanes J, Pe’er D, Tata PR, Rawlins E, Regev A, Reyfman P, Samakovlis C, Schultze J, Shalek A, Shepherd D, Spence J, Teichmann S, Theis F, Tsankov A, van den Berge M, von Papen M, Whitsett J, Zaragosi LE, Angelov A, Bals R, Bartholomäus A, Becker A, Bezdan D, Bonifacio E, Bork P, Clavel T, Colme-Tatche M, Diefenbach A, Dilthey A, Fischer N, Förstner K, Frick J-S, Gagneur J, Goesmann A, Hain T, Hummel M, Janssen S, Kalinowski J, Kallies R, Kehr B, Keller A, Kim-Hellmuth S, Klein C, Kohlbacher O, Korbel JO, Kurth I, Landthaler M, Li Y, Ludwig K, Makarewicz O, Marz M, McHardy A, Mertes C, Nöthen M, Nürnberg P, Ohler U, Ossowski S, Overmann J, Peter S, Pfeffer K, Poetsch AR, Pühler A, Rajewsky N, Ralser M, Rieß O, Ripke S, Nunes da Rocha U, Rosenstiel P, Saliba A-E, Sander LE, Sawitzki B, Schiffer P, Schulte E-C, Schultze JL, Sczyrba A, Stegle O, Stoye J, Theis F, Vehreschild J, Vogel J, von Kleist M, Walker A, Walter J, Wieczorek D, Ziebuhr J. 2020. Longitudinal Multi-omics Analyses Identify Responses of Megakaryocytes, Erythroid Cells, and Plasmablasts as Hallmarks of Severe COVID-19. Immunity 53:1296–1314.e9.
Liu C, Martins AJ, Lau WW, Rachmaninoff N, Chen J, Imberti L, Mostaghimi D, Fink DL, Burbelo PD, Dobbs K, Delmonte OM, Bansal N, Failla L, Sottini A, Quiros-Roldan E, Han KL, Sellers BA, Cheung F, Sparks R, Chun T-W, Moir S, Lionakis MS, Rossi C, Su HC, Kuhns DB, Cohen JI, Notarangelo LD, Tsang JS, Abers MS, Apps R, Bosticardo M, Milanez-Almeida P, Mulè MP, Shaw E, Zhang Y, Castelli F, Muiesan ML, Tomasoni G, Scolari F, Tucci A. 2021. Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19. Cell 184:1836–1857.e22.
Arunachalam PS, Wimmers F, Mok CKP, Perera RAPM, Scott M, Hagan T, Sigal N, Feng Y, Bristow L, Tak-Yin Tsang O, Wagh D, Coller J, Pellegrini KL, Kazmin D, Alaaeddine G, Leung WS, Chan JMC, Chik TSH, Choi CYC, Huerta C, Paine McCullough M, Lv H, Anderson E, Edupuganti S, Upadhyay AA, Bosinger SE, Maecker HT, Khatri P, Rouphael N, Peiris M, Pulendran B. 2020. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 369:1210–1220.
Yang AC, Kern F, Losada PM, Agam MR, Maat CA, Schmartz GP, Fehlmann T, Stein JA, Schaum N, Lee DP, Calcuttawala K, Vest RT, Berdnik D, Lu N, Hahn O, Gate D, McNerney MW, Channappa D, Cobos I, Ludwig N, Schulz-Schaeffer WJ, Keller A, Wyss-Coray T. 2021. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 595:565–571.
Ju B, Zhang Q, Ge X, Wang R, Yu J, Shan S, Zhou B, Song S, Tang X, Yu J, Ge J, Lan J, Yuan J, Wang H, Zhao J, Zhang S, Wang Y, Shi X, Liu L, Wang X, Zhang Z, Zhang L. 2020. Potent human neutralizing antibodies elicited by SARS-CoV-2 infection. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.21.990770.
Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, Wheeler DK, Sette A, Peters B. 2019. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Research 47:D339–D343.
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, Tummino TA, Hüttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Meyer B, Roesch F, Vallet T, Mac Kain A, Miorin L, Moreno E, Naing ZZC, Zhou Y, Peng S, Shi Y, Zhang Z, Shen W, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Lyu J, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Rakesh R, Liu X, Rosenthal SB, Calviello L, Venkataramanan S, Liboy-Lugo J, Lin Y, Huang X-P, Liu Y, Wankowicz SA, Bohn M, Safari M, Ugur FS, Koh C, Savar NS, Tran QD, Shengjuler D, Fletcher SJ, O’Neal MC, Cai Y, Chang JCJ, Broadhurst DJ, Klippsten S, Sharp PP, Wenzell NA, Kuzuoglu-Ozturk D, Wang H-Y, Trenker R, Young JM, Cavero DA, Hiatt J, Roth TL, Rathore U, Subramanian A, Noack J, Hubert M, Stroud RM, Frankel AD, Rosenberg OS, Verba KA, Agard DA, Ott M, Emerman M, Jura N, von Zastrow M, Verdin E, Ashworth A, Schwartz O, d’Enfert C, Mukherjee S, Jacobson M, Malik HS, Fujimori DG, Ideker T, Craik CS, Floor SN, Fraser JS, Gross JD, Sali A, Roth BL, Ruggero D, Taunton J, Kortemme T, Beltrao P, Vignuzzi M, García-Sastre A, Shokat KM, Shoichet BK, Krogan NJ. 2020. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583:459–468.
Li J, Guo M, Tian X, Liu C, Wang X, Yang X, Wu P, Xiao Z, Qu Y, Yin Y, Fu J, Zhu Z, Liu Z, Peng C, Zhu T, Liang Q. 2020. Virus-host interactome and proteomic survey of PMBCs from COVID-19 patients reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.31.019216.
Navratil V, Lionnard L, Longhi S, Hardwick JM, Combet C, Aouacheria A. 2020. The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) envelope (E) protein harbors a conserved BH3-like sequence. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.04.09.033522.
198.
Overmyer KA, Shishkova E, Miller IJ, Balnis J, Bernstein MN, Peters-Clarke TM, Meyer JG, Quan Q, Muehlbauer LK, Trujillo EA, He Y, Chopra A, Chieng HC, Tiwari A, Judson MA, Paulson B, Brademan DR, Zhu Y, Serrano LR, Linke V, Drake LA, Adam AP, Schwartz BS, Singer HA, Swanson S, Mosher DF, Stewart R, Coon JJ, Jaitovich A. 2021. Large-Scale Multi-omic Analysis of COVID-19 Severity. Cell Systems 12:23–40.e7.
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X. 2020. Crystal structure of the 2019-nCoV spike receptor-binding domain bound with the ACE2 receptor. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.19.956235.
Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L, Chen H-D, Chen J, Luo Y, Guo H, Jiang R-D, Liu M-Q, Chen Y, Shen X-R, Wang X, Zheng X-S, Zhao K, Chen Q-J, Deng F, Liu L-L, Yan B, Zhan F-X, Wang Y-Y, Xiao G-F, Shi Z-L. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273.
Lu J, du Plessis L, Liu Z, Hill V, Kang M, Lin H, Sun J, François S, Kraemer MUG, Faria NR, McCrone JT, Peng J, Xiong Q, Yuan R, Zeng L, Zhou P, Liang C, Yi L, Liu J, Xiao J, Hu J, Liu T, Ma W, Li W, Su J, Zheng H, Peng B, Fang S, Su W, Li K, Sun R, Bai R, Tang X, Liang M, Quick J, Song T, Rambaut A, Loman N, Raghwani J, Pybus OG, Ke C. 2020. Genomic Epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell 181:997–1003.e9.
Fauver JR, Petrone ME, Hodcroft EB, Shioda K, Ehrlich HY, Watts AG, Vogels CBF, Brito AF, Alpert T, Muyombwe A, Razeq J, Downing R, Cheemarla NR, Wyllie AL, Kalinich CC, Ott IM, Quick J, Loman NJ, Neugebauer KM, Greninger AL, Jerome KR, Roychoudhury P, Xie H, Shrestha L, Huang M-L, Pitzer VE, Iwasaki A, Omer SB, Khan K, Bogoch II, Martinello RA, Foxman EF, Landry ML, Neher RA, Ko AI, Grubaugh ND. 2020. Coast-to-Coast Spread of SARS-CoV-2 during the Early Epidemic in the United States. Cell 181:990–996.e5.
217.
Gonzalez-Reiche AS, Hernandez MM, Sullivan MJ, Ciferri B, Alshammary H, Obla A, Fabre S, Kleiner G, Polanco J, Khan Z, Alburquerque B, van de Guchte A, Dutta J, Francoeur N, Melo BS, Oussenko I, Deikus G, Soto J, Sridhar SH, Wang Y-C, Twyman K, Kasarskis A, Altman DR, Smith M, Sebra R, Aberg J, Krammer F, García-Sastre A, Luksza M, Patel G, Paniz-Mondolfi A, Gitman M, Sordillo EM, Simon V, van Bakel H. 2020. Introductions and early spread of SARS-CoV-2 in the New York City area. Science eabc1917.
218.
Gudbjartsson DF, Helgason A, Jonsson H, Magnusson OT, Melsted P, Norddahl GL, Saemundsdottir J, Sigurdsson A, Sulem P, Agustsdottir AB, Eiriksdottir B, Fridriksdottir R, Gardarsdottir EE, Georgsson G, Gretarsdottir OS, Gudmundsson KR, Gunnarsdottir TR, Gylfason A, Holm H, Jensson BO, Jonasdottir A, Jonsson F, Josefsdottir KS, Kristjansson T, Magnusdottir DN, le Roux L, Sigmundsdottir G, Sveinbjornsson G, Sveinsdottir KE, Sveinsdottir M, Thorarensen EA, Thorbjornsson B, Löve A, Masson G, Jonsdottir I, Möller AD, Gudnason T, Kristinsson KG, Thorsteinsdottir U, Stefansson K. 2020. Spread of SARS-CoV-2 in the Icelandic Population. New England Journal of Medicine 382:2302–2315.
Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B, Hastie KM, Parker MD, Partridge DG, Evans CM, Freeman TM, de Silva TI, McDanal C, Perez LG, Tang H, Moon-Walker A, Whelan SP, LaBranche CC, Saphire EO, Montefiori DC, Angyal A, Brown RL, Carrilero L, Green LR, Groves DC, Johnson KJ, Keeley AJ, Lindsey BB, Parsons PJ, Raza M, Rowland-Jones S, Smith N, Tucker RM, Wang D, Wyles MD. 2020. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 182:812–827.e19.
223.
Yurkovetskiy L, Wang X, Pascal KE, Tomkins-Tinch C, Nyalile TP, Wang Y, Baum A, Diehl WE, Dauphin A, Carbone C, Veinotte K, Egri SB, Schaffner SF, Lemieux JE, Munro JB, Rafique A, Barve A, Sabeti PC, Kyratsous CA, Dudkina NV, Shen K, Luban J. 2020. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 183:739–751.e8.
224.
Plante JA, Liu Y, Liu J, Xia H, Johnson BA, Lokugamage KG, Zhang X, Muruato AE, Zou J, Fontes-Garfias CR, Mirchandani D, Scharton D, Bilello JP, Ku Z, An Z, Kalveram B, Freiberg AN, Menachery VD, Xie X, Plante KS, Weaver SC, Shi P-Y. 2020. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 592:116–121.
225.
Thomson EC, Rosen LE, Shepherd JG, Spreafico R, da Silva Filipe A, Wojcechowskyj JA, Davis C, Piccoli L, Pascall DJ, Dillen J, Lytras S, Czudnochowski N, Shah R, Meury M, Jesudason N, De Marco A, Li K, Bassi J, O’Toole A, Pinto D, Colquhoun RM, Culap K, Jackson B, Zatta F, Rambaut A, Jaconi S, Sreenu VB, Nix J, Zhang I, Jarrett RF, Glass WG, Beltramello M, Nomikou K, Pizzuto M, Tong L, Cameroni E, Croll TI, Johnson N, Di Iulio J, Wickenhagen A, Ceschi A, Harbison AM, Mair D, Ferrari P, Smollett K, Sallusto F, Carmichael S, Garzoni C, Nichols J, Galli M, Hughes J, Riva A, Ho A, Schiuma M, Semple MG, Openshaw PJM, Fadda E, Baillie JK, Chodera JD, Rihn SJ, Lycett SJ, Virgin HW, Telenti A, Corti D, Robertson DL, Snell G. 2021. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 184:1171–1187.e20.
Volz E, Mishra S, Chand M, Barrett JC, Johnson R, Geidelberg L, Hinsley WR, Laydon DJ, Dabrera G, O’Toole Á, Amato R, Ragonnet-Cronin M, Harrison I, Jackson B, Ariani CV, Boyd O, Loman NJ, McCrone JT, Gonçalves S, Jorgensen D, Myers R, Hill V, Jackson DK, Gaythorpe K, Groves N, Sillitoe J, Kwiatkowski DP, Flaxman S, Ratmann O, Bhatt S, Hopkins S, Gandy A, Rambaut A, Ferguson NM, The COVID-19 Genomics UK (COG-UK) consortium. 2021. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.12.30.20249034.
van Doremalen N, Bushmaker T, Morris DH, Holbrook MG, Gamble A, Williamson BN, Tamin A, Harcourt JL, Thornburg NJ, Gerber SI, Lloyd-Smith JO, de Wit E, Munster VJ. 2020. Aerosol and Surface Stability of SARS-CoV-2 as Compared with SARS-CoV-1. New England Journal of Medicine 382:1564–1567.
Lednicky JA, Lauzardo M, Fan ZH, Jutla A, Tilly TB, Gangwar M, Usmani M, Shankar SN, Mohamed K, Eiguren-Fernandez A, Stephenson CJ, Alam MdM, Elbadry MA, Loeb JC, Subramaniam K, Waltzek TB, Cherabuddi K, Morris JG Jr., Wu C-Y. 2020. Viable SARS-CoV-2 in the air of a hospital room with COVID-19 patients. International Journal of Infectious Diseases 100:476–482.
265.
Santarpia JL, Herrera VL, Rivera DN, Ratnesar-Shumate S, Reid StP, Denton PW, Martens JWS, Fang Y, Conoan N, Callahan MV, Lawler JV, Brett-Major DM, Lowe JJ. 2020. The Infectious Nature of Patient-Generated SARS-CoV-2 Aerosol. Cold Spring Harbor Laboratory.
Comber L, O Murchu E, Drummond L, Carty PG, Walsh KA, De Gascun CF, Connolly MA, Smith SM, O'Neill M, Ryan M, Harrington P. 2020. Airborne transmission of SARS‐CoV‐2 via aerosols. Rev Med Virol 31.
269.
Bourouiba L. 2020. Turbulent Gas Clouds and Respiratory Pathogen Emissions. JAMA https://doi.org/10.1001/jama.2020.4756.
He X, Lau EHY, Wu P, Deng X, Wang J, Hao X, Lau YC, Wong JY, Guan Y, Tan X, Mo X, Chen Y, Liao B, Chen W, Hu F, Zhang Q, Zhong M, Wu Y, Zhao L, Zhang F, Cowling BJ, Li F, Leung GM. 2020. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nature Medicine 26:672–675.
Arons MM, Hatfield KM, Reddy SC, Kimball A, James A, Jacobs JR, Taylor J, Spicer K, Bardossy AC, Oakley LP, Tanwar S, Dyal JW, Harney J, Chisty Z, Bell JM, Methner M, Paul P, Carlson CM, McLaughlin HP, Thornburg N, Tong S, Tamin A, Tao Y, Uehara A, Harcourt J, Clark S, Brostrom-Smith C, Page LC, Kay M, Lewis J, Montgomery P, Stone ND, Clark TA, Honein MA, Duchin JS, Jernigan JA. 2020. Presymptomatic SARS-CoV-2 Infections and Transmission in a Skilled Nursing Facility. New England Journal of Medicine 382:2081–2090.
Zhang P, Tian F, Wan Y, Cai J, Qian Z, Wu R, Zhang Y, Zhang S, Li H, Li M, Trevathan E, Lin H. 2020. A Cohort of SARS-CoV-2 Infected Asymptomatic and Pre-Symptomatic Contacts from COVID-19 Contact Tracing in Hubei Province, China: Short-Term Outcomes. SSRN Electronic Journal https://doi.org/10.2139/ssrn.3678556.
Ladhani SN, Chow JY, Janarthanan R, Fok J, Crawley-Boevey E, Vusirikala A, Fernandez E, Perez MS, Tang S, Dun-Campbell K, Evans EW-, Bell A, Patel B, Amin-Chowdhury Z, Aiano F, Paranthaman K, Ma T, Saavedra-Campos M, Myers R, Ellis J, Lackenby A, Gopal R, Patel M, Brown C, Chand M, Brown K, Ramsay ME, Hopkins S, Shetty N, Zambon M. 2020. Investigation of SARS-CoV-2 outbreaks in six care homes in London, April 2020. EClinicalMedicine 26:100533.
Lavezzo E, Franchin E, Ciavarella C, Cuomo-Dannenburg G, Barzon L, Del Vecchio C, Rossi L, Manganelli R, Loregian A, Navarin N, Abate D, Sciro M, Merigliano S, De Canale E, Vanuzzo MC, Besutti V, Saluzzo F, Onelia F, Pacenti M, Parisi SG, Carretta G, Donato D, Flor L, Cocchio S, Masi G, Sperduti A, Cattarino L, Salvador R, Nicoletti M, Caldart F, Castelli G, Nieddu E, Labella B, Fava L, Drigo M, Gaythorpe KAM, Brazzale AR, Toppo S, Trevisan M, Baldo V, Donnelly CA, Ferguson NM, Dorigatti I, Crisanti A, Imperial College COVID-19 Response Team, Imperial College COVID-19 Response Team. 2020. Suppression of a SARS-CoV-2 outbreak in the Italian municipality of Vo’. Nature 584:425–429.
Grewelle R, De Leo G. 2020. Estimating the Global Infection Fatality Rate of COVID-19. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.05.11.20098780.
Ma S, Zhang J, Zeng M, Yun Q, Guo W, Zheng Y, Zhao S, Wang MH, Yang Z. 2020. Epidemiological parameters of coronavirus disease 2019: a pooled analysis of publicly reported individual data of 1155 cases from seven countries. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.21.20040329.
303.
Majumder M, Mandl KD. 2020. Early Transmissibility Assessment of a Novel Coronavirus in Wuhan, China. SSRN Electronic Journal https://doi.org/10.2139/ssrn.3524675.
304.
Liu T, Hu J, Xiao J, He G, Kang M, Rong Z, Lin L, Zhong H, Huang Q, Deng A, Zeng W, Tan X, Zeng S, Zhu Z, Li J, Gong D, Wan D, Chen S, Guo L, Li Y, Sun L, Liang W, Song T, He J, Ma W. 2020. Time-varying transmission dynamics of Novel Coronavirus Pneumonia in China. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.01.25.919787.
Shen M, Peng Z, Xiao Y, Zhang L. 2020. Modelling the epidemic trend of the 2019 novel coronavirus outbreak in China. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.01.23.916726.
309.
Read JM, Bridgen JRE, Cummings DAT, Ho A, Jewell CP. 2020. Novel coronavirus 2019-nCoV: early estimation of epidemiological parameters and epidemic predictions. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.01.23.20018549.
310.
Roques L, Klein E, Papaïx J, Sar A, Soubeyrand S. 2020. Using early data to estimate the actual infection fatality ratio from COVID-19 in France. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.22.20040915.
Kucharski AJ, Russell TW, Diamond C, Liu Y, Edmunds J, Funk S, Eggo RM, Sun F, Jit M, Munday JD, Davies N, Gimma A, van Zandvoort K, Gibbs H, Hellewell J, Jarvis CI, Clifford S, Quilty BJ, Bosse NI, Abbott S, Klepac P, Flasche S. 2020. Early dynamics of transmission and control of COVID-19: a mathematical modelling study. The Lancet Infectious Diseases 20:553–558.
313.
Sahafizadeh E, Sartoli S. 2020. Estimating the reproduction number of COVID-19 in Iran using epidemic modeling. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.20.20038422.
314.
Flaxman S, Mishra S, Gandy A, Unwin H, Coupland H, Mellan T, Zhu H, Berah T, Eaton J, Perez Guzman P, Schmit N, Cilloni L, Ainslie K, Baguelin M, Blake I, Boonyasiri A, Boyd O, Cattarino L, Ciavarella C, Cooper L, Cucunuba Perez Z, Cuomo-Dannenburg G, Dighe A, Djaafara A, Dorigatti I, Van Elsland S, Fitzjohn R, Fu H, Gaythorpe K, Geidelberg L, Grassly N, Green W, Hallett T, Hamlet A, Hinsley W, Jeffrey B, Jorgensen D, Knock E, Laydon D, Nedjati Gilani G, Nouvellet P, Parag K, Siveroni I, Thompson H, Verity R, Volz E, Walters C, Wang H, Wang Y, Watson O, Winskill P, Xi X, Whittaker C, Walker P, Ghani A, Donnelly C, Riley S, Okell L, Vollmer M, Ferguson N, Bhatt S. 2020. Report 13: Estimating the number of infections and the impact of non-pharmaceutical interventions on COVID-19 in 11 European countries. Imperial College London.
Washington NL, Gangavarapu K, Zeller M, Bolze A, Cirulli ET, Barrett KMS, Larsen BB, Anderson C, White S, Cassens T, Jacobs S, Levan G, Nguyen J, Ramirez JM III, Rivera-Garcia C, Sandoval E, Wang X, Wong D, Spencer E, Robles-Sikisaka R, Kurzban E, Hughes LD, Deng X, Wang C, Servellita V, Valentine H, De Hoff P, Seaver P, Sathe S, Gietzen K, Sickler B, Antico J, Hoon K, Liu J, Harding A, Bakhtar O, Basler T, Austin B, Isaksson M, Febbo PG, Becker D, Laurent M, McDonald E, Yeo GW, Knight R, Laurent LC, de Feo E, Worobey M, Chiu C, Suchard MA, Lu JT, Lee W, Andersen KG. 2021. Genomic epidemiology identifies emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the United States. Cold Spring Harbor Laboratory.
317.
Davies NG, Abbott S, Barnard RC, Jarvis CI, Kucharski AJ, Munday JD, Pearson CAB, Russell TW, Tully DC, Washburne AD, Wenseleers T, Gimma A, Waites W, Wong KLM, van Zandvoort K, Silverman JD, Diaz-Ordaz K, Keogh R, Eggo RM, Funk S, Jit M, Atkins KE, Edmunds WJ. 2021. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 372.
EpiForecasts and the CMMID Covid working group. Covid-19: Temporal variation in transmission during the COVID-19 outbreak. https://epiforecasts.io/covid/. Retrieved 27 July 2020.
324.
Systrom K, Vladeck T, Krieger M. Rt COVID-19. https://rt.live/. Retrieved 27 July 2020.
Riemersma KK, Haddock LA III, Wilson NA, Minor N, Eickhoff J, Grogan BE, Kita-Yarbro A, Halfmann PJ, Segaloff HE, Kocharian A, Florek KR, Westergaard R, Bateman A, Jeppson GE, Kawaoka Y, O’Connor DH, Friedrich TC, Grande KM. 2021. Shedding of Infectious SARS-CoV-2 Despite Vaccination. Cold Spring Harbor Laboratory.
van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJM, Wolthers KC, Wertheim-van Dillen PME, Kaandorp J, Spaargaren J, Berkhout B. 2004. Identification of a new human coronavirus. Nature Medicine 10:368–373.
Bloom JD, Chan YA, Baric RS, Bjorkman PJ, Cobey S, Deverman BE, Fisman DN, Gupta R, Iwasaki A, Lipsitch M, Medzhitov R, Neher RA, Nielsen R, Patterson N, Stearns T, van Nimwegen E, Worobey M, Relman DA. 2021. Investigate the origins of COVID-19. Science 372:694–694.
Hosie MJ, Hofmann-Lehmann R, Hartmann K, Egberink H, Truyen U, Addie DD, Belák S, Boucraut-Baralon C, Frymus T, Lloret A, Lutz H, Marsilio F, Pennisi MG, Tasker S, Thiry E, Möstl K. 2021. Anthropogenic Infection of Cats during the 2020 COVID-19 Pandemic. Viruses 13:185.
389.
Sit THC, Brackman CJ, Ip SM, Tam KWS, Law PYT, To EMW, Yu VYT, Sims LD, Tsang DNC, Chu DKW, Perera RAPM, Poon LLM, Peiris M. 2020. Infection of dogs with SARS-CoV-2. Nature 586:776–778.
390.
Decaro N, Vaccari G, Lorusso A, Lorusso E, De Sabato L, Patterson EI, Di Bartolo I, Hughes GL, Teodori L, Desario C, Colitti B, Ricci D, Buonavoglia D, Rosati S, Martella V, Cammà C, Agrimi U, Elia G. 2021. Possible Human-to-Dog Transmission of SARS-CoV-2, Italy, 2020. Emerg Infect Dis 27:1981–1984.
391.
Patterson EI, Elia G, Grassi A, Giordano A, Desario C, Medardo M, Smith SL, Anderson ER, Prince T, Patterson GT, Lorusso E, Lucente MS, Lanave G, Lauzi S, Bonfanti U, Stranieri A, Martella V, Solari Basano F, Barrs VR, Radford AD, Agrimi U, Hughes GL, Paltrinieri S, Decaro N. 2020. Evidence of exposure to SARS-CoV-2 in cats and dogs from households in Italy. Nat Commun 11.
Bosco-Lauth AM, Walker A, Guilbert L, Porter S, Hartwig A, McVicker E, Bielefeldt-Ohmann H, Bowen RA. 2021. Susceptibility of livestock to SARS-CoV-2 infection. Emerging Microbes & Infections 10:2199–2201.
Villanueva-Saz S, Giner J, Fernández A, Lacasta D, Ortín A, Ramos JJ, Ferrer LM, Ruiz de Arcaute M, Tobajas AP, Pérez MD, Verde M, Marteles D, Hurtado-Guerrero R, Pardo J, Santiago L, González-Ramírez AM, Macías-León J, García-García A, Taleb V, Lira-Navarrete E, Paño-Pardo JR, Ruíz H. 2021. Absence of SARS-CoV-2 Antibodies in Natural Environment Exposure in Sheep in Close Contact with Humans. Animals 11:1984.
Oreshkova N, Molenaar RJ, Vreman S, Harders F, Oude Munnink BB, Hakze-van der Honing RW, Gerhards N, Tolsma P, Bouwstra R, Sikkema RS, Tacken MG, de Rooij MM, Weesendorp E, Engelsma MY, Bruschke CJ, Smit LA, Koopmans M, van der Poel WH, Stegeman A. 2020. SARS-CoV-2 infection in farmed minks, the Netherlands, April and May 2020. Eurosurveillance 25.
Conceicao C, Thakur N, Human S, Kelly JT, Logan L, Bialy D, Bhat S, Stevenson-Leggett P, Zagrajek AK, Hollinghurst P, Varga M, Tsirigoti C, Tully M, Chiu C, Moffat K, Silesian AP, Hammond JA, Maier HJ, Bickerton E, Shelton H, Dietrich I, Graham SC, Bailey D. 2020. The SARS-CoV-2 Spike protein has a broad tropism for mammalian ACE2 proteins. PLoS Biol 18:e3001016.
, Niemi MEK, Karjalainen J, Liao RG, Neale BM, Daly M, Ganna A, Pathak GA, Andrews SJ, Kanai M, Veerapen K, Fernandez-Cadenas I, Schulte EC, Striano P, Marttila M, Minica C, Marouli E, Karim MA, Wendt FR, Savage J, Sloofman L, Butler-Laporte G, Kim H-N, Kanoni S, Okada Y, Byun J, Han Y, Uddin MJ, Smith GD, Willer CJ, Buxbaum JD, Mehtonen J, Finucane H, Cordioli M, Martin AR, Zhou W, Pasaniuc B, Julienne H, Aschard H, Shi H, Yengo L, Polimanti R, Ghoussaini M, Schwartzentruber J, Dunham I, Chwialkowska K, Francescatto M, Trankiem A, Balaconis MK, Davis L, Lee S, Priest J, Renieri A, Sankaran VG, van Heel D, Deelen P, Richards JB, Nakanishi T, Biesecker L, Kerchberger VE, Baillie JK, Mari F, Bernasconi A, Baillie SC, Canakoglu A, Wolford B, Faucon A, Dutta AK, Schurmann C, Harry E, Birney E, Nguyen H, Nasir J, Kaunisto M, Solomonson M, Dueker N, Vadgama N, Limou S, Rahmouni S, Mbarek H, Darwish D, Uddin MM, Albertos R, Pérez-Tur J, Li R, Folkersen L, Moltke I, Koelling N, Teumer A, Kousathanas A, Utrilla A, Verdugo RA, Zárate R, Medina-Gómez C, Gómez-Cabrero D, Carnero-Montoro E, Cadilla CL, Moreno-Estrada A, Garmendia A, Moya L, Sedaghati-Khayat B, Boua PR, Favé M-J, Francioli L, Lemaçon A, Migeotte I, Patel S, Varnai R, Szentpeteri JL, Sipeky C, Colombo F, von Hohenstaufen K, Lio P, Vallerga C, Wang Q, Tanigawa Y, Im H, Han C, Song H, Lim J, Lee Y, Kim S, Im S, Atanasovska B, Ahmad HF, Boer C, Jansen P, Franke L, Kaja E, Pasko D, Kennis-Szilagyi I, Kornilov SA, Prijatelj V, Prokić I, Sivanadhan I, Perumal S, Esmaeeli S, Pearson NM, Auton A, Shelton JF, Shastri AJ, Filshtein-Sonmez T, Coker D, Symons A, Esparza-Gordillo J, Aslibekyan S, O’Connell J, Ye C, Weldon CH, Perera M, O’Leary K, Tuck M, O’Brien T, Meltzer D, O’Donnell P, Nutescu E, Yang G, Alarcon C, Herrmann S, Mazurek S, Banagan J, Hamidi Z, Barbour A, Raffat N, Moreno D, Friedman P, Ferwerda B, van de Beek D, Brouwer MC, Vlaar APJ, Wiersinga WJ, Posthuma D, Tissink E, Koos Zwinderman AH, Uffelmann E, van Agtmael M, Algera AG, van Baarle F, Bax D, Beudel M, Jan Bogaard H, Bomers M, Bonta PI, Bos L, Botta M, de Brabander J, de Bree G, de Bruin S, Bugiani M, Bulle E, Chouchane O, Cloherty A, Dongelmans D, Elbers P, Fleuren L, Geerlings S, Geerts B, Geijtenbeek T, Girbes A, Goorhuis B, Grobusch MP, Hafkamp F, Hagens L, Hamann J, Harris V, Hemke R, Hermans SM, Heunks L, Hollmann M, Horn J, Hovius JW, de Jong MD, Koning R, van Mourik N, Nellen J, Nossent EJ, Paulus F, Peters E, van der Poll T, Preckel B, Prins JM, Raasveld J, Reijnders T, Schinkel M, Schultz MJ, Schuurman A, Sigaloff K, Smit M, Stijnis CS, Stilma W, Teunissen C, Thoral P, Tsonas A, van der Valk M, Veelo D, de Vries H, van Vugt M, Wouters D, Minnaar RP, Kromhout A, van Uffelen KWJ, Wolterman RA, Roberts G, Park D, Ball CA, Coignet M, McCurdy S, Knight S, Partha R, Rhead B, Zhang M, Berkowitz N, Gaddis M, Noto K, Ruiz L, Pavlovic M, Hong EL, Rand K, Girshick A, Guturu H, Baltzell AH, Rahmouni S, Guntz J, Beguin Y, Pigazzini S, Nkambule L, Bouysran Y, Busson A, Peyrassol X, Wilkin F, Pichon B, Smits G, Vandernoot I, Goffard J-C, Georges M, Moutschen M, Misset B, Darcis G, Guiot J, Jadot L, Azarzar S, Dellot P, Gofflot S, Claassen S, Bertrand A, Parzibut G, Clarinval M, Moermans C, Malaise O, El Kandoussi K, Thonon R, Huynen P, Mesdagh A, Melo S, Jacques N, Di Valentin E, Giroule F, Collignon A, Radermecker C, Lebrun M, Perée H, Latour S, Barada O, Sanchez J, Josse C, Boujemla B, Meunier M, Mariavelle E, Anania S, Gazon H, Juszczak D, Fadeur M, Camby S, Meuris C, Thys M, Jacques J, Henket M, Léonard P, Frippiat F, Giot J-B, Sauvage A-S, Von Frenckell C, Mni M, Wéry M, Staderoli A, Belhaj Y, Lambermont B, Morrison DR, Mooser V, Forgetta V, Li R, Ghosh B, Laurent L, Belisle A, Henry D, Abdullah T, Adeleye O, Mamlouk N, Kimchi N, Afrasiabi Z, Rezk N, Vulesevic B, Bouab M, Guzman C, Petitjean L, Tselios C, Xue X, Afilalo J, Afilalo M, Oliveira M, Brenner B, Brassard N, Durand M, Schurr E, Lepage P, Ragoussis J, Auld D, Chassé M, Kaufmann DE, Lathrop GM, Adra D, Davis LK, Cox NJ, Below JE, Sealock JM, Faucon AB, Shuey MM, Polikowsky HG, Petty LE, Shaw DM, Chen H-H, Zhu W, Ludwig KU, Schröder J, Maj C, Rolker S, Nöthen MM, Fazaal J, Keitel V, Jensen B-EO, Feldt T, Kurth I, Marx N, Dreher M, Pink I, Cornberg M, Illig T, Lehmann C, Schommers P, Augustin M, Rybniker J, Knopp L, Eggermann T, Volland S, Altmüller J, Berger MM, Brenner T, Hinney A, Witzke O, Bals R, Herr C, Ludwig N, Walter J, Fuchsberger C, Pattaro C, De Grandi A, Pramstaller P, Emmert D, Melotti R, Foco L, Mascalzoni D, Gögele M, Domingues F, Hicks A, Gignoux CR, Wicks SJ, Crooks K, Barnes KC, Daya M, Shortt J, Rafaels N, Chavan S, Goldstein DB, Kiryluk K, Sengupta S, Chung W, Reilly MP, Khan A, Wang C, Povysil G, Bhardwaj N, Gharavi AG, Ionita-Laza I, Shang N, O’Byrne SM, Nandakumar R, Menon A, So YS, Hod E, Pendrick D, Kim H-N, Park S-K, Kim H-L, Kang CK, Lee H-J, Song K-H, Jae Yoon K, Paik N-J, Seok W, Yoon H, Joo E-J, Chang Y, Ryu S, Park WB, Su Park J, Un Park K, Ham SY, Jung J, Kim ES, Kim HB, Ellinghaus D, Degenhardt F, Cáceres M, Juzenas S, Lenz TL, Albillos A, Julià A, Heidecker B, Garcia F, Kurth F, Tran F, Hanses F, Zoller H, Holter JC, Fernández J, Sander LE, Rosenstiel P, Koehler P, de Cid R, Asselta R, Schreiber S, Hehr U, Prati D, Baselli G, Valenti L, Bujanda L, Banales JM, Duga S, D’Amato M, Romero-Gómez M, Buti M, Invernizzi P, Franke A, Hov JR, Karlsen TH, Folseraas T, Maya-Miles D, Teles A, Azuure C, Wacker EM, Uellendahl-Werth F, ElAbd H, Arora J, Lerga-Jaso J, Wienbrandt L, Rühlemann MC, Wendorff M, Vadla MS, Lenning OB, Özer O, Myhre R, Raychaudhuri S, Tanck A, Gassner C, Hemmrich-Stanisak G, Kässens J, Figuera Basso ME, Schulzky M, Wittig M, Braun N, Wesse T, Albrecht W, Yi X, Ortiz AB, Chercoles AG, Ruiz A, Mantovani A, Holten AR, Mayer A, Cherubini A, Protti A, Aghemo A, Gerussi A, Ramirez A, Braun A, Barreira A, Lleo A, Kildal AB, Glück A, Nolla AC, Latiano A, Dyrhol-Riise AM, Muscatello A, Voza A, Rando-Segura A, Solier A, Karina B, Cortes B, Mateos B, Nafria-Jimenez B, Schaefer B, Bellinghausen C, Ferrando C, Quereda C, Skurk C, Thibeault C, Spinner CD, Lange C, Hu C, Cappadona C, Bianco C, Sancho C, Lihaug Hoff DA, Galimberti D, Jiménez D, Pestaña D, Toapanta D, Azzolini E, Scarpini E, Helbig ET, Urrechaga E, Paraboschi EM, Pontali E, Reverter E, Navas E, Arana E, Sánchez FG, Ceriotti F, Malvestiti F, Mesonero F, Pezzoli G, Lamorte G, Neb H, My I, Hernández I, de Rojas I, Galván-Femenia I, Heyckendorf J, Rybniker J, Badia JR, Schneider J, Goikoetxea J, Kraft J, Müller KE, Gaede KI, Garcia-Etxebarria K, Tonby K, Heggelund L, Izquierdo-Sanchez L, Sumoy L, Lippert LJ, Terranova L, Garbarino L, Téllez L, Roade L, Ostadreza M, Intxausti M, Kogevinas M, Gutiérrez-Stampa MA, Vehreschild MJGT, Marquié M, Castoldi M, Cecconi M, Boada M, Seilmaier MJ, Mazzocco M, Rodríguez-Gandía M, Ayo NI, Blay N, Martínez N, Cornely OA, Palmieri O, Tentorio P, Rodrigues PM, España PP, Hoffmann P, Bacher P, Suwalski P, de Pablo R, Nieto R, Badalamenti S, Ciesek S, Bombace S, Wilfling S, Brunak S, Heilmann-Heimbach S, Ripke S, Bahmer T, Landmesser U, Protzer U, Rimoldi V, Skogen V, Andrade V, Moreno V, Poller W, Farre X, Wang X, Khodamoradi Y, Karadeniz Z, de Salazar A, Palom A, Garcia-Fernandez A-E, Blanco-Grau A, Zanella A, Bandera A, Nebel A, Biondi A, Caballero-Garralda A, Gori A, Lind A, Fracanzani AL, Peschuck A, Pesenti A, de la Horra C, Milani C, Paccapelo C, Angelini C, Cea C, Muñiz-Diaz E, Sandoval E, Calderón EJ, Solligård E, Aziz F, Martinelli-Boneschi F, Peyvandi F, Blasi F, Medrano FJ, Rodriguez-Frias F, Müller F, Grasselli G, Costantino G, Cardamone G, Foti G, Matullo G, Kurihara H, Afset JE, Damås JK, Ampuero J, Martín J, Erdmann J, Bergan J, Goerg S, Ferrusquía-Acosta J, Quero JH, Delgado J, Guerrero JM, Risnes K, Bettini LR, Moreira L, Gustad LT, Santoro L, Scudeller L, Riveiro-Barciela M, Schaefer M, Carrabba M, Valsecchi MG, Hernandez-Tejero M, Acosta-Herrera M, D’Angiò M, Baldini M, Cazzaniga M, Ciccarelli M, Bocciolone M, Miozzo M, Chueca N, Montano N, Faverio P, Preatoni P, Bonfanti P, Omodei P, Castro P, Ferrer R, Gualtierotti R, Gallego-Durán R, Morilla R, Haider S, Marsal S, Aneli S, Pelusi S, Bosari S, Aliberti S, Dudman S, Zheng T, Pumarola T, Cejudo TG, Monzani V, Friaza V, Peter W, Dopazo X, Duga S, May S, Grimsrud MM, Gudbjartsson DF, Stefansson K, Sulem P, Sveinbjornsson G, Melsted P, Norddahl G, Swerford Moore KH, Thorsteinsdottir U, Holm H, Alarcón-Riquelme ME, Bernardo D, Martínez-Bueno M, Rello SR, Magi R, Milani L, Metspalu A, Laisk T, Läll K, Lepamets M, Esko T, Reimann E, Naaber P, Laane E, Pesukova J, Peterson P, Kisand K, Tabri J, Allos R, Hensen K, Starkopf J, Ringmets I, Tamm A, Kallaste A, Alavere H, Metsalu K, Puusepp M, Kristiansson K, Koskelainen S, Perola M, Donner K, Kivinen K, Palotie A, Palotie A, Rivolta C, Bochud P-Y, Bibert S, Boillat N, Nussle SG, Albrich W, Quinodoz M, Kamdar D, Suh N, Neofytos D, Erard V, Voide C, Bochud PY, Rivolta C, Bibert S, Quinodoz M, Kamdar D, Neofytos D, Erard V, Voide C, Friolet R, Vollenweider P, Pagani JL, Oddo M, zu Bentrup FM, Conen A, Clerc O, Marchetti O, Guillet A, Guyat-Jacques C, Foucras S, Rime M, Chassot J, Jaquet M, Viollet RM, Lannepoudenx Y, Portopena L, Desgranges F, Filippidis P, Guéry B, Haefliger D, Kampouri EE, Manuel O, Munting A, Papadimitriou-Olivgeris M, Regina J, Rochat-Stettler L, Suttels V, Tadini E, Tschopp J, Van Singer M, Viala B, Boillat-Blanco N, Brahier T, Hügli O, Meuwly JY, Pantet O, Nussle SG, Bochud M, D’Acremont V, Younes SE, Albrich WC, Suh N, Cerny A, O’Mahony L, von Mering C, Bochud PY, Frischknecht M, Kleger G-R, Filipovic M, Kahlert CR, Wozniak H, Negro TR, Pugin J, Bouras K, Knapp C, Egger T, Perret A, Montillier P, di Bartolomeo C, Barda B, de Cid R, Carreras A, Moreno V, Galván-Femenía I, Blay N, Farré X, Sumoy L, Cortés B, Mercader JM, Guindo-Martinez M, Torrents D, Garcia-Aymerich J, Castaño-Vinyals G, Dobaño C, Gori M, Renieri A, Mondelli MU, Castelli F, Vaghi M, Rusconi S, Montagnani F, Bargagli E, Franchi F, Mazzei MA, Cantarini L, Tacconi D, Feri M, Scala R, Spargi G, Nencioni C, Bandini M, Caldarelli GP, Spagnesi M, Canaccini A, Ognibene A, D’Arminio Monforte A, Girardis M, Antinori A, Francisci D, Schiaroli E, Scotton PG, Panese S, Scaggiante R, Monica MD, Capasso M, Fiorentino G, Castori M, Aucella F, Di Biagio A, Masucci L, Valente S, Mandalà M, Zucchi P, Giannattasio F, Coviello DA, Mussini C, Bosio G, Tavecchia L, Crotti L, Rizzi M, La Rovere MT, Sarzi-Braga S, Bussotti M, Ravaglia S, Artuso R, Perrella A, Romani D, Bergomi P, Catena E, Vincenti A, Ferri C, Grassi D, Pessina G, Tumbarello M, Di Pietro M, Sabrina R, Luchi S, Barbieri C, Acquilini D, Andreucci E, Paciosi F, Segala FV, Tiseo G, Falcone M, Lista M, Poscente M, De Vivo O, Petrocelli P, Guarnaccia A, Baroni S, Perticaroli V, Furini S, Dei S, Benetti E, Picchiotti N, Sanarico M, Ceri S, Pinoli P, Raimondi F, Biscarini F, Stella A, Bergomi M, Zguro K, Capitani K, Tanfoni M, Fallerini C, Daga S, Baldassarri M, Fava F, Frullanti E, Valentino F, Doddato G, Giliberti A, Tita R, Amitrano S, Bruttini M, Croci S, Meloni I, Mencarelli MA, Lo Rizzo C, Pinto AM, Beligni G, Tommasi A, Di Sarno L, Palmieri M, Carriero ML, Alaverdian D, Iuso N, Inchingolo G, Busani S, Bruno R, Vecchia M, Belli MA, Mantovani S, Ludovisi S, Quiros-Roldan E, Antoni MD, Zanella I, Siano M, Emiliozzi A, Fabbiani M, Rossetti B, Zanelli G, Bergantini L, D’Alessandro M, Cameli P, Bennet D, Anedda F, Marcantonio S, Scolletta S, Guerrini S, Conticini E, Frediani B, Spertilli C, Donati A, Guidelli L, Corridi M, Croci L, Piacentini P, Desanctis E, Cappelli S, Verzuri A, Anemoli V, Pancrazi A, Lorubbio M, Merlini E, Miraglia FG, Venturelli S, Cossarizza A, Vergori A, Gabrieli A, Riva A, Paciosi F, Andretta F, Gatti F, Parisi SG, Baratti S, Piscopo C, Russo R, Andolfo I, Iolascon A, Carella M, Merla G, Squeo GM, Raggi P, Marciano C, Perna R, Bassetti M, Sanguinetti M, Giorli A, Salerni L, Parravicini P, Menatti E, Trotta T, Coiro G, Lena F, Martinelli E, Mancarella S, Gabbi C, Maggiolo F, Ripamonti D, Bachetti T, Suardi C, Parati G, Bottà G, Di Domenico P, Rancan I, Bianchi F, Colombo R, van Heel DA, Hunt KA, Trembath RC, Huang QQ, Martin HC, Mason D, Trivedi B, Wright J, Finer S, Griffiths CJ, Akhtar S, Anwar M, Arciero E, Ashraf S, Breen G, Chung R, Curtis CJ, Chowdhury M, Colligan G, Deloukas P, Durham C, Finer S, Griffiths C, Huang QQ, Hurles M, Hunt KA, Hussain S, Islam K, Khan A, Khan A, Lavery C, Lee SH, Lerner R, MacArthur D, MacLaughlin B, Martin H, Mason D, Miah S, Newman B, Safa N, Tahmasebi F, Trembath RC, Trivedi B, van Heel DA, Wright J, Smith AV, Boughton AP, Li KW, LeFaive J, Annis A, Jannes CE, Krieger JE, Pereira AC, Velho M, Marques E, Lima IR, Tada MT, Valino K, McCarthy M, Rosenberger C, Lee JE, Chang D, Hammer C, Hunkapiller J, Mahajan A, Pendergrass S, Sucheston-Campbell L, Yaspan B, Lee HS, Shin E, Jang HY, Kim S, Kym S, Kim Y-S, Jeong H, Kwon KT, Kim S-W, Kim JY, Jang YR, Kim H ah, Lee JY, Lee JE, Lee S, Choe K-W, Kang YM, Jee SH, Jung KJ, Parikh V, Ashley E, Wheeler M, Rivas M, Bustamante C, Pinksy B, Febbo P, Farh K, Schroth GP, deSouza F, Dalton K, Christle J, Deboever C, Szalma S, Tanigawa Y, Rubinacci S, Delaneau O, Gorzynski J, de Jong H, Sutton S, Youlton N, Joshi R, Jimenez-Morales D, Hughes C, Amar D, Ioannidis A, Hershman S, Kirillova A, Seo K, Huang Y, Shoura M, Hammond N, Watson N, Raja A, Huang C, Sahoo M, Wang H, Zhen J, Rakitko A, Ilinsky V, Yermakovich D, Popov I, Chernitsov A, Kovalenko E, Krasnenko A, Plotnikov N, Stetsenko I, Kim A, Cirulli ET, Schiabor Barrett KM, Bolze A, White S, Washington NL, Lu JT, Riffle S, Tanudjaja F, Wang X, Ramirez JM III, Leonetti N, Sandoval E, Neveux I, Dabe S, Grzymski JJ, Esteban Miñano JI, Aguirre LA, López-Collazo E, de la Mata Pazos M, Cerrato L, Folkersen L, Lozano-Rodríguez R, Avendaño-Ortiz J, Arcos VT, Montalbán-Hernández KM, Quiroga JV, Pascual-Iglesias A, Maroun-Eid C, Martín-Quirós A, Namkoong H, Okada Y, Imoto S, Katayama K, Fukunaga K, Kitagawa Y, Sato T, Hasegawa N, Kumanogoh A, Kimura A, Ai M, Tokunaga K, Kanai T, Miyano S, Ogawa S, Edahiro R, Sonehara K, Shirai Y, Kanai M, Ishii M, Kabata H, Masaki K, Kamata H, Ikemura S, Chubachi S, Okamori S, Terai H, Tanaka H, Morita A, Lee H, Asakura T, Sasaki J, Morisaki H, Uwamino Y, Nanki K, Mikami Y, Tomono K, Kato K, Matsuda F, Takahashi M, Hizawa N, Takeda Y, Hirata H, Shiroyama T, Miyawaki S, Suzuki K, Maeda Y, Nii T, Noda Y, Niitsu T, Adachi Y, Enomoto T, Amiya S, Hara R, Takahashi K, Anzai T, Hasegawa T, Ito S, Koike R, Endo A, Uchimura Y, Miyazaki Y, Honda T, Tateishi T, Tohda S, Ichimura N, Sonobe K, Sassa C, Nakajima J, Nannya Y, Omae Y, Takahashi K, Harada N, Hiki M, Takagi H, Nakamura A, Tagaya E, Kawana M, Arimura K, Ishiguro T, Takayanagi N, Isono T, Takaku Y, Takano K, Anan R, Nakajima Y, Nakano Y, Nishio K, Ueda S, Hayashi R, Tateno H, Hase I, Yoshida S, Suzuki S, Mitamura K, Saito F, Ueda T, Azuma M, Nagasaki T, Yasui Y, Hasegawa Y, Mutoh Y, Yoshiyama T, Shoko T, Kojima M, Adachi T, Ishikawa M, Takahashi K, Watanabe K, Manabe T, Ito F, Fukui T, Funatsu Y, Koh H, Hirai Y, Kawashima H, Narita A, Niwa K, Sekikawa Y, Saito F, Yoshiya K, Yoshihara T, Suzuki Y, Nakayama S, Masuzawa K, Nishi K, Nishitsuji M, Tani M, Inoue T, Hirano T, Kobayashi K, Miyazawa N, Kimura Y, Sado R, Ogura T, Kitamura H, Murohashi K, Nakachi I, Baba R, Arai D, Fuke S, Saito H, Kuwahara N, Fujiwara A, Okada T, Baba T, Noda J, Mashimo S, Yagi K, Shiomi T, Hashiguchi M, Odani T, Mochimaru T, Oyamada Y, Mori N, Izumi N, Nagata K, Taki R, Murakami K, Yamada M, Sugiura H, Hayashi K, Shimizu T, Gon Y, Fujitani S, Tsuchida T, Yoshida T, Kagaya T, Kita T, Sakagami S, Kimizuka Y, Kawana A, Nakamura Y, Ishikura H, Takata T, Kikuchi T, Taniyama D, Nakamura M, Kodama N, Kaneyama Y, Maeda S, Nagasaki Y, Okamoto M, Ishihara S, Ito A, Chihara Y, Takeuchi M, Onoi K, Hashimoto N, Wakahara K, Ando A, Masuda M, Wakabayashi A, Watanabe H, Sageshima H, Nakada T-A, Abe R, Shimada T, Kawamura K, Ichikado K, Nishiyama K, Yamasaki M, Hashimoto S, Kusaka Y, Ohba T, Isogai S, Takada M, Kanda H, Komase Y, Sano F, Asano K, Oguma T, Harada M, Takahashi T, Shibusawa T, Abe S, Kono Y, Togashi Y, Izumo T, Inomata M, Awano N, Ogawa S, Ogata T, Ishihara S, Kanehiro A, Ozaki S, Fuchimoto Y, Kitagawa Y, Yoshida S, Ogura S, Nishiyama K, Yoshida K, Beppu S, Fukuyama S, Eriguchi Y, Yonekawa A, Inoue Y, Yamagata K, Chiba S, Narumoto O, Nagai H, Ooshima N, Motegi M, Sagara H, Tanaka A, Ohta S, Shibata Y, Tanino Y, Sato Y, Yamada Y, Hashino T, Shinoki M, Iwagoe H, Imamura T, Umeda A, Shimada H, Endo M, Hayashi S, Takahashi M, Nakano S, Yatomi M, Maeno T, Ishii T, Utsugi M, Ono A, Kanaoka K, Ihara S, Komuta K, Franke L, Boezen M, Claringbould A, Lopera E, Warmerdam R, Vonk JudithM, van Blokland I, Lanting P, Ori APS, Obeidat M, Hernández Cordero AI, Sin DD, Bossé Y, Joubert P, Hao K, Nickle D, Timens W, van den Berge M, Feng Y-CA, Mercader J, Weiss ST, Karlson EW, Smoller JW, Murphy SN, Meigs JB, Woolley AE, Green RC, Perez EF, Wolford B, Zöllner S, Wang J, Beck A, Sloofman LG, Ascolillo S, Sebra RP, Collins BL, Levy T, Sealfon SC, Jordan DM, Thompson RC, Gettler K, Chaudhary K, Belbin GM, Preuss M, Hoggart C, Choi S, Underwood SJ, Salib I, Britvan B, Keller K, Tang L, Peruggia M, Hiester LL, Niblo K, Aksentijevich A, Labkowsky A, Karp A, Zlatopolsky M, Zyndorf M, Charney AW, Beckmann ND, Schadt EE, Abul-Husn NS, Cho JH, Itan Y, Kenny EE, Loos RJF, Nadkarni GN, Do R, O’Reilly P, Huckins LM, Ferreira MAR, Abecasis GR, Leader JB, Cantor MN, Justice AE, Carey DJ, Chittoor G, Josyula NS, Kosmicki JA, Horowitz JE, Baras A, Gass MC, Yadav A, Mirshahi T, Jan Hottenga J, Bartels M, de Geus EJC, Nivard MG, Verma A, Ritchie MD, Rader D, Li B, Verma SS, Lucas A, Bradford Y, Zara F, Salpietro V, Scala M, Iacomino M, Scudieri P, Bocciardi R, Minetti C, Riva A, Vari MS, Rahier J-F, Giorgio E, Carli D, Louis E, Bulik CM, Landén M, Brusco A, Ferrero GB, Madia F, Fundín B, Ismail SI, Saad C, Al-Sarraj Y, Badji RM, Al-Muftah W, Al Thani A, Afifi N, Klovins J, Rovite V, Rescenko R, Peculis R, Ustinova M, Zeberg H, Frithiof R, Hultström M, Lipcsey M, Johnson R, Geschwind DH, Freimer N, Butte MJ, Geschwind DH, Pasaniuc B, Ding Y, Chiu A, Chang TS, Boutros P, Moutsianas L, Caulfield MJ, Scott RH, Walker S, Stuckey A, Odhams CA, Rhodes D, Fowler T, Rendon A, Chan G, Arumugam P, Karczewski KJ, Wilson DJ, Spencer CA, Crook DW, Wyllie DH, O’Connell AM, Atkinson EG, Kanai M, Tsuo K, Baya N, Turley P, Gupta R, Walters RK, Palmer DS, Sarma G, Solomonson M, Cheng N, Lu W, Churchhouse C, Goldstein JI, King D, Seed C, Daly MJ, Neale BM, Bryant S, Satterstrom FK, Band G, Earle SG, Lin S-K, Arning N, Armstrong J, Rudkin JK, Callier S, Bryant S, Cusick C, Soranzo N, Zhao JH, Danesh J, Di Angelantonio E, Butterworth AS, Sun YV, Huffman JE, Cho K, O’Donnell CJ, Tsao P, Gaziano JM, Peloso G, Ho Y-L, Mian M, Scaggiante F, Chang X, Glessner JR, Hakonarson H, McGuigan PJ, Prockter Moore LS, Vizcaychipi MP, Hall K, Campbell A, Nichol A, Ward G, Page VJ, Semple MG, Adeniji K, Agranoff D, Agwuh K, Ail D, Aldera EL, Alegria A, Angus B, Ashish A, Atkinson D, Bari S, Barlow G, Barnass S, Barrett N, Bassford C, Basude S, Baxter D, Beadsworth M, Bernatoniene J, Berridge J, Best N, Bothma P, Chadwick D, Brittain-Long R, Bulteel N, Burden T, Burtenshaw A, Caruth V, Chambler D, Chee N, Child J, Chukkambotla S, Clark T, Collini P, Cosgrove C, Cupitt J, Cutino-Moguel M-T, Dark P, Dawson C, Dervisevic S, Donnison P, Douthwaite S, Drummond A, DuRand I, Dushianthan A, Dyer T, Evans C, Eziefula C, Fegan C, Finn A, Fullerton D, Garg S, Garg A, Gkrania-Klotsas E, Godden J, Goldsmith A, Graham C, Hardy E, Hartshorn S, Harvey D, Havalda P, Hawcutt DB, Hobrok M, Hodgson L, Hormis A, Jacobs M, Jain S, Jennings P, Kaliappan A, Kasipandian V, Kegg S, Kelsey M, Kendall J, Kerrison C, Kerslake I, Koch O, Koduri G, Koshy G, Laha S, Laird S, Larkin S, Leiner T, Lillie P, Limb J, Linnett V, Little J, Lyttle M, MacMahon M, MacNaughton E, Mankregod R, Masson H, Matovu E, McCullough K, McEwen R, Meda M, Mills GH, Minton J, Ward K, Mirfenderesky M, Mohandas K, Mok Q, Moon J, Moore E, Morgan P, Morris C, Mortimore K, Moses S, Mpenge M, Mulla R, Murphy M, Nagel M, Nagarajan T, Nelson M, O’Shea MK, Otahal I, Ostermann M, Pais M, Panchatsharam S, Papakonstantinou D, Paraiso H, Patel B, Pattison N, Pepperell J, Peters M, Phull M, Pintus S, Pooni JS, Post F, Price D, Prout R, Rae N, Reschreiter H, Reynolds T, Richardson N, Roberts M, Roberts D, Rose A, Rousseau G, Ryan B, Saluja T, Shah A, Shanmuga P, Sharma A, Shawcross A, Sizer J, Shankar-Hari M, Smith R, Snelson C, Spittle N, Staines N, Stambach T, Stewart R, Subudhi P, Szakmany T, Tatham K, Thomas J, Thompson C, Thompson R, Tridente A, Tupper-Carey D, Twagira M, Ustianowski A, Vallotton N, Vincent-Smith L, Visuvanathan S, Vuylsteke A, Waddy S, Wake R, Walden A, Welters I, Whitehouse T, Whittaker P, Whittington A, Papineni P, Wijesinghe M, Williams M, Wilson L, Cole S, Winchester S, Wiselka M, Wolverson A, Wooton DG, Workman A, Yates B, Young P, Beale R, Bretherick AD, Clohisey S, Fourman MH, Furniss J, Gountouna E, Grimes G, Haley C, Harrison D, Hayward C, Keating S, Klaric L, Klenerman P, Law A, Meynert AM, Millar J, Pairo-Castineira E, Parkinson N, Ponting CP, Porteous DJ, Rawlik K, Richmond A, Rowan K, Russell CD, Scott RH, Shen X, Shih B, Tenesa A, Vitart V, Wang B, Wilson JF, Wu Y, Yang J, Yang Z, Zechner M, Zhai R, Zheng C, Norman L, Pius R, Drake TM, Fairfield CJ, Knight SR, Mclean KA, Murphy D, Shaw CA, Dalton J, Girvan M, Saviciute E, Roberts S, Harrison J, Marsh L, Connor M, Halpin S, Jackson C, Gamble C, Leeming G, Law A, Wham M, Hendry R, Scott-Brown J, Begg C, Hinds C, Wai Ho AY, Horby PW, Knight J, Ling L, Maslove D, McAuley D, Montgomery H, Nichol A, Openshaw PJM, Semple MG, Shankar-Hari M, Summers C, Walsh T, Armstrong L, Bates H, Dooks E, Farquhar F, Hairsine B, McParland C, Packham S, Alldis Z, Astin-Chamberlain R, Bibi F, Biddle J, Blow S, Bolton M, Borra C, Bowles R, Burton M, Choudhury Y, Collier D, Cox A, Easthope A, Ebano P, Fotiadis S, Gurasashvili J, Halls R, Hartridge P, Kallon D, Kassam J, Lancoma-Malcolm I, Matharu M, May P, Mitchelmore O, Newman T, Patel M, Pheby J, Pinzuti I, Prime Z, Prysyazhna O, Shiel J, Taylor M, Tierney C, Wood S, Zak A, Zongo O, Forsey M, Kaliappan A, Nicholson A, Riches J, Vertue M, Wasson C, Finn S, Green J, Collins E, King B, Grauslyte L, Hussain M, Phull M, Pogreban T, Rosaroso L, Salciute E, Franke G, Wong J, George A, Akeroyd L, Bano S, Bromley M, Gurr L, Lawton T, Morgan J, Sellick K, Warren D, Wilkinson B, McGowan J, Ledgard C, Stacey A, Pye K, Bellwood R, Bentley M, Hobrok M, Loosley R, McGuinness H, Tench H, Wolf-Roberts R, Gibson S, Lyle A, McNeela F, Radhakrishnan J, Hughes A, Ali A, Brady M, Dale S, Dance A, Gledhill L, Greig J, Hanson K, Holdroyd K, Home M, Kelly D, Kitson R, Matapure L, Melia D, Mellor S, Nortcliffe T, Pinnell J, Robinson M, Shaw L, Shaw R, Thomis L, Wilson A, Wood T, Bayo L-A, Merwaha E, Ishaq T, Hanley S, Antcliffe D, Banach D, Brett S, Coghlan P, Fernandez Z, Gordon A, Rojo R, Arias SS, Templeton M, Jha R, Krishnamurthy V, Lim L, Bi R, Scholefield B, Ashton L, Williams A, Cheyne C, Saunderson A, Allan A, Anderson F, Kaye C, Liew J, Medhora J, Scott T, Trumper E, Botello A, Polgarova P, Stroud K, Meaney E, Jones M, Ng A, Agrawal S, Pathan N, White D, Daubney E, Elston K, Parker R, Reddy A, Turner-Bone I, Wilding L, Harding P, Jacob R, Jones C, Denmade C, Croft M, White I, Lim L, Griffin D, Muchenje N, Mupudzi M, Partridge R, Conyngham J-A, Thomas R, Wright M, Corral MA, Bastion V, Clarke D, David B, Kent H, Lorusso R, Lubimbi G, Murdoch S, Penacerrada M, Thomas A, Valentine J, Vochin A, Wulandari R, Djeugam B, Dawson J, Garrioch S, Tolson M, Aldridge J, de Almeida Martins LG, Carungcong J, Beavis S, Dale K, Gascoyne R, Hawes J, Pritchard K, Stevenson L, Whileman A, Cowley A, Highgate J, Crawley R, Crew A, Cunningham M, Daniels A, Harrison L, Hope S, Inweregbu K, Jones S, Lancaster N, Matthews J, Nicholson A, Wray G, Benham L, Bradshaw Z, Brown J, Caswell M, Cupitt J, Melling S, Preston S, Slawson N, Stoddard E, Warden S, Combes E, Joefield T, Monnery S, Beech V, Trotman S, Hopkins B, Scriven J, Thrasyvoulou L, Willis H, Anderson S, Birch J, Collins E, Hammerton K, O’Leary R, Abernathy C, Foster L, Gratrix A, Martinson V, Parkinson P, Stones E, Carbral-Ortega L, Kapoor R, Loader D, Castle K, Brandwood C, Smith L, Clark R, Birchall K, Kolakaluri L, Baines D, Sukumaran A, Mapfunde I, Meredith M, Morris L, Ryan L, Clark A, Sampson J, Peters C, Dent M, Langley M, Ashraf S, Wei S, Andrew A, Chablani M, Kirkby A, Netherton K, Bates M, Dasgin J, Gill J, Nilsson A, Scriven J, Apetri E, Basikolo C, Blackledge B, Catlow L, Charles B, Dark P, Doonan R, Harris J, Harvey A, Horner D, Knowles K, Lee S, Lomas D, Lyons C, Marsden T, McLaughlan D, McMorrow L, Pendlebury J, Perez J, Poulaka M, Proudfoot N, Slaughter M, Slevin K, Taylor M, Thomas V, Walker D, Michael A, Collis M, Clark M, Coulding M, Jude E, McCormick J, Mercer O, Potla D, Rehman H, Savill H, Turner V, Davey M, Golden D, Seaman R, Hunt J, Dearden J, Dobson E, Mulcahy M, Munt S, O’Connor G, Philbin J, Rishton C, Tully R, Winnard S, Cagova L, Fofano A, Garner L, Holcombe H, Mepham S, Mitchell AM, Mwaura L, Praman K, Vuylsteke A, Zamikula J, Bercades G, Brealey D, Hass I, MacCallum N, Martir G, Raith E, Reyes A, Smyth D, Taylor A, Hughes RA, Thomas H, Rees A, Duskova M, Phipps J, Brooks S, Edwards M, Alexander P, Allen S, Bradley-Potts J, Brantwood C, Egan J, Felton T, Padden G, Ward L, Moss S, Glasgow S, Beesley K, Board S, Kubisz-Pudelko A, Lewis A, Perry J, Pippard L, Wood D, Buckley C, Brown A, Gregory J, O’Connell S, Smith T, Belagodu Z, Fuller B, Gherman A, Olufuwa O, Paramsothy R, Stuart C, Oakley N, Kamundi C, Tyl D, Collins K, Silva P, Taylor J, King L, Coates C, Crowley M, Wakefield P, Beadle J, Johnson L, Sargeant J, Anderson M, Jardine C, Williams D, Parris V, Quaid S, Watson E, Melville J, Naisbitt J, Joseph R, Lazo M, Walton O, Neal A, Hill M, Kannan T, Wild L, Allan E, Darlington K, Davies F, Easton J, Kumar S, Lean R, Menzies D, Pugh R, Qiu X, Davies L, Williams H, Scanlon J, Davies G, Mackay C, Lewis J, Rees S, Coetzee S, Gales A, Otahal I, Raj M, Sell C, Langton H, Prout R, Watters M, Novis C, Arbane G, Bociek A, Campos S, Grau N, Jones TO, Lim R, Marotti M, Ostermann M, Shankar-Hari M, Whitton C, Barron A, Collins C, Kaul S, Passmore H, Prendergast C, Reed A, Rogers P, Shokkar R, Woodruff M, Middleton H, Polgar O, Nolan C, Thwaites V, Mahay K, Sri-Chandana C, Scherewode J, Stephenson L, Marsh S, Bancroft H, Bellamy M, Carmody M, Daglish J, Moore F, Rhodes J, Sangombe M, Kadiri S, Scriven J, Ayers A, Harrison W, North J, Cavazza A, Cockrell M, Corcoran E, Depante M, Finney C, Jerome E, McPhail M, Nayak M, Noble H, O’Reilly K, Pappa E, Saha R, Saha S, Smith J, Knighton A, Gill M, Paul P, Ratnam V, Shelton S, Wynter I, Baptista D, Crowe R, Fernandes R, Herdman-Grant R, Joseph A, Loveridge A, McKenley I, Morino E, Naranjo A, Simms R, Sollesta K, Swain A, Venkatesh H, Khera J, Fox J, Barber R, Hewitt C, Hilldrith A, Jackson-Lawrence K, Shepardson S, Wills M, Butler S, Tavares S, Cunningham A, Hindale J, Arif S, George L, Twiss S, Wright D, Holland M, Keenan N, Lyons M, Wassall H, Marsh C, Mahenthran M, Carter E, Kong T, Adanini O, Bhatia N, Msiska M, Mew L, Mwaura E, Stewart R, Williams F, Wren L, Sutherland S-B, Battle C, Brinkworth E, Harford R, Murphy C, Newey L, Rees T, Williams M, Arnold S, Brealey D, Hardy J, Houlden H, Moncur E, Raith E, Tariq A, Tucci A, Convery K, Fottrell-Gould D, Hudig L, Keshet-Price J, Randell G, Stammers K, Abdelrazik M, Bakthavatsalam D, Elhassan M, Ganesan A, Haldeos A, Moreno-Cuesta J, Purohit D, Vincent R, Xavier K, Rohit K, Alasdair F, Saleem M, David C, Jenkins S, Lamond Z, Wall A, Yates B, Reynolds J, Campbell H, Thompsom M, Dodds S, Duffy S, Butcher D, O’Sullivan S, Butterworth-Cowin N, Deacon B, Hibbert M, Pothecary C, Tetla D, Woodford C, Durga L, Kennard-Holden G, de Gordoa LO-R, Peasgood E, Phillips C, Skinner D, Gaylard J, Mullan D, Newman J, Davies E, Roche L, Sathe S, Brimfield L, Daly Z, Pogson D, Rose S, Collins A, Khaliq W, Gude ET, Allen L, Beranova E, Crisp N, Deery J, Hazelton T, Knight A, Price C, Tilbey S, Turki S, Turney S, Giles J, Booth S, Bell G, English K, Katary A, Wilcox L, Campbell R, Clarke N, Whiteside J, Mascarenhas M, Donaldson A, Matheson J, Barrett F, O’Hara M, O’Keefe L, Bradley C, Collier D, Hormis A, Walker R, Maynard V, Patel T, Smith M, Chukkambotla S, Kazi A, Hartley J, Dykes J, Hijazi M, Keith S, Khan M, Ryan-Smith J, Springle P, Thomas J, Truman N, Saad S, Coleman D, Fine C, Matt R, Gay B, Dalziel J, Ali S, Goodchild D, Harling R, Bhatterjee R, Goddard W, Davison C, Duberly S, Hargreaves J, Bolton R, Laha S, Verlander M, Williams A, Blackman H, Creagh-Brown B, Donlon S, Michalak-Glinska N, Mtuwa S, Pristopan V, Salberg A, Smith E, Stone S, Piercy C, Verula J, Burda D, Montaser R, Harden L, Mayangao I, Marriott C, Bradley P, Harris C, Cooper J, Finch C, Liderth S, Quinn A, Waddington N, Fidler K, Tagliavini E, Donnelly K, Abel L, Brett M, Digby B, Gemmell L, Hornsby J, MacGoey P, O’Neil P, Price R, Rodden N, Rooney K, Sundaram R, Thomson N, Flanagan R, Hughes G, Latham S, McKenna E, Anderson J, Hull R, Rhead K, Branney D, Frankham J, Pitts S, White N, Cristiano D, Dormand N, Farzad Z, Gummadi M, Liyanage K, Patel BV, Salmi S, Sloane G, Thwaites V, Varghese M, Zborowski AC, Bean S, Burt K, Spivey M, Eastgate-Jackson C, Filipe H, Martin D, Maharajh A, Garcia SM, De Neef M, Deacon B, Lynch C, Pothecary C, Roche L, Howe GS, Singh J, Turner K, Ellis H, Stroud N, Cherian S, Cutler S, Heron AE, Roynon-Reed A, Szakmany T, Williams G, Richards O, Cheema Y, Ahmad N, Barker J, Bauchmuller K, Bird S, Cawthron K, Harrington K, Jackson Y, Kibutu F, Lenagh B, Masuko S, Mills GH, Raithatha A, Wiles M, Willson J, Newell H, Lye A, Nwafor L, Jarman C, Rowland-Jones S, Foote D, Cole J, Thompson R, Watson J, Hesseldon L, Macharia I, Chetam L, Smith J, Ford A, Anderson S, Birchall K, Housley K, Walker S, Milner L, Hanratty H, Trower H, Phillips P, Oxspring S, Donne B, Bevan E, Martin J, Trodd D, Watson G, Brown CW, Bunni L, Jennings C, Latif M, Marshall R, Subramanian G, Bandla N, Gellamucho M, Davies M, Thompson C, Trim F, Eapen B, Ahmed C, Baines B, Clamp S, Colley J, Haq R, Hayes A, Hulme J, Hussain S, Joseph S, Kumar R, Maqsood Z, Purewal M, Chandler B, Elliott K, Mallinson J, Turnbull A, Dent K, Horsley E, Akhtar MN, Pearson S, Potoczna D, Spencer S, Blakemore H, Borislavova B, Faulkner B, Gendall E, Goff E, Hayes K, Thomas M, Worner R, Smith K, Stephens D, Delgado CC, Dawson D, Ding L, Durrant G, Ezeobu O, Farnell-Ward S, Harrison A, Kanu R, Leaver S, Maccacari E, Manna S, Saluzzio RP, Queiroz J, Samakomva T, Sicat C, Texeira J, Da Gloria EF, Lisboa A, Rawlins J, Mathew J, Kinch A, Hurt WJ, Shah N, Clark V, Thanasi M, Yun N, Patel K, Crickmore V, Debreceni G, Wilkins J, Nicol L, Burn I, Hambrook G, Manso K, Penn R, Shanmugasundaram P, Tebbutt J, Thornton D, Rostron A, Roy A, Woods L, Cornell S, Wakinshaw F, Rogerson K, Jarmain J, Anderson P, Archer K, Austin K, Davis C, Durie A, Kelsall O, Thrush J, Vigurs C, Wild L, Wood H-L, Tranter H, Harrison A, Cowley N, McAlindon M, Burtenshaw A, Digby S, Low E, Morgan A, Cother N, Rankin T, Clayton S, McCurdy A, Allibone S, Mary-Genetu R, Kasipandian V, Patel A, Mac A, Murphy A, Mahjoob P, Nazari R, Worsley L, Fagan A, Mohamed Ali IA, Beaumont K, Blunt M, Coton Z, Curgenven H, Elsaadany M, Fernandes K, Ally SM, Rangarajan H, Sarathy V, Selvanayagam S, Vedage D, White M, Fernandez-Roman J, Hamilton DO, Johnson E, Johnston B, Martinez ML, Mulla S, Shaw D, Waite AAC, Waugh V, Welters ID, Williams K, Bemand T, Black E, Rosa AD, Howle R, Jhanji S, Baikady RR, Tatham KC, Thomas B, Halkes M, Mercer P, Thornton L, West J, Baird T, Ruddy J, Reece-Anthony R, Birt M, Cowton A, Kay A, Kent M, Potts K, Wilkinson A, Naylor S, Brown E, Clark M, Purvis S, Cole J, Davies M, Davies R, Duffin D, Hill H, Player B, Thomas E, Williams A, Beith CM, Black K, Clements S, Morrison A, Strachan D, Taylor M, Clarkson M, D’Sylva S, Norman K, Coventry T, Fowler S, MacMahon M, McGregor A, Brady A, Chan R, Little J, McIvor S, Prady H, Whittle H, Mathew B, Clapham M, Harper R, Poultney U, Rice P, Smith T, Mutch R, Baird Y, Butler A, Chadbourn I, Folkes L, Fox H, Gardner A, Gomez R, Hobden G, Hodgson L, King K, Margarson M, Martindale T, Meadows E, Raynard D, Thirlwall Y, Helm D, Margalef J, Greer S, Shuker K, Tridente A, Smuts S, Duffield J, Smith O, Mallon L, Claire W, Birkinshaw I, Carter J, Howard K, Ingham J, Joy R, Pearson H, Roche S, Scott Z, Knights E, Price A, Thomas A, Thorpe C, Abraheem A, Bamford P, Cawley K, Dunmore C, Faulkner M, Girach R, Jeffrey H, Jones R, London E, Nagra I, Nasir F, Sainsbury H, Smedley C, Khade R, Sundar A, Tsinaslanidis G, Behan T, Burnett C, Hatton J, Heeney E, Mitra A, Newton M, Pollard R, Stead R, Birch J, Bough L, Goodsell J, Tutton R, Williams P, Williams S, Winter-Goodwin B, Auld F, Donnachie J, Edmond I, Prentice L, Runciman N, Salutous D, Symon L, Todd A, Turner P, Short A, Sweeney L, Murdoch E, Senaratne D, Burns K, Higham A, Anderson T, Hawcutt D, O’Malley L, Rad L, Rogers N, Saunderson P, Allison KS, Afolabi D, Whitbread J, Jones D, Dore R, Lankester L, Nikitas N, Wells C, Stowe B, Spencer K, Cathcart S, Duffy K, Puxty A, Puxty K, Turner L, Ireland J, Semple G, Barry P, Hilltout P, Evitts J, Tyler A, Waldron J, Irvine V, Shelley B, Akinkugbe O, Bamford A, Beech E, Belfield H, Bell M, Davies C, Jones GAL, McHugh T, Meghari H, O’Neill L, Peters MJ, Ray S, Tomas AL, Easthope A, Gorman C, Gupta A, Timlick E, Brady R, Bonner S, Hugill K, Jones J, Liggett S, Bashyal A, Davidson N, Hutton P, McKechnie S, Wilson J, Flint N, Rekha P, Hales D, Cruz C, Pattison N, Gopal S, Harris N, Lake V, Metherell S, Radford E, Clement I, Patel B, Gulati A, Hays C, Webster K, Hudson A, Webster A, Stephenson E, McCormack L, Slater V, Nixon R, Hanson H, Fearby M, Kelly S, Bridgett V, Robinson P, Almaden-Boyle C, Austin P, Cabrelli L, Cole S, Casey M, Chapman S, Whyte C, Brayne A, Fisher E, Hunt J, Jackson P, Kaye D, Love N, Parkin J, Tuckey V, van Koutrik L, Carter S, Andrew B, Findlay L, Adams K, Bruce M, Connolly K, Duncan T, T.-Michael H, Lindergard G, Hey S, Fox C, Alfonso J, Durrans LJ, Guerin J, Blackledge B, Harris J, Hruska M, Eltayeb A, Lamb T, Hodgkiss T, Cooper L, Rothwell J, Dennis C, McGregor A, Parris V, Srikaran S, Sukha A, Davies K, O’Brien L, Omar Z, Otahal I, Perkins E, Lewis T, Sutherland I, Brooke H, Buckley S, Suarez JC, Charlesworth R, Hansson K, Norris J, Poole A, Rose A, Sandhu R, Sloan B, Smithson E, Thirumaran M, Wagstaff V, Metcalfe A, Camsooksai J, Humphrey C, Jenkins S, Reschreiter H, Wadams B, DeAth Y, Adams C, Agasou A, Arden T, Bowes A, Boyle P, Beekes M, Button H, Capps N, Carnahan M, Carter A, Childs D, Donaldson D, Hard K, Hurford F, Hussain Y, Javaid A, Jones J, Jose S, Leigh M, Martin T, Millward H, Motherwell N, Rikunenko R, Stickley J, Summers J, Ting L, Tivenan H, Tonks L, Wilcox R, Bokhari M, Linnett V, Lucas R, McCormick W, Ritzema J, Sanderson A, Wild H, Baxter N, Henderson S, Kennedy-Hay S, McParland C, Rooney L, Sim M, McCreath G, Brunton M, Caterson J, Coles H, Frise M, Rai SG, Jacques N, Keating L, Tilney E, Bartley S, Bhuie P, Downes C, Holding K, Riches K, Hilton M, Hayman M, Subramanian D, Daniel P, Zitter L, Benyon S, Marriott S, Park L, Keenan S, Gordon E, Quinn H, Baines K, Andrew G, Baillie JK, Barclay L, Callaghan M, Campbell R, Clark S, Hope D, Marshall L, McCulloch C, Briton K, Singleton J, Birch S, Higham A, Simpson K, Craig J, Demetriou C, Eckbad C, Hierons S, Howie L, Mitchard S, Ramos L, Serrano-Ruiz A, White K, Kelly F, Amin V, Anastasescu E, Anumakonda V, Karthik K, Kausar R, Reid K, Smith J, Imeson-Wood J, Bellini A, Bryant J, Mayer A, Pickard A, Roe N, Sowter J, Howlett A, Criste K, Cusack R, Golder K, Golding H, Jones O, Leggett S, Male M, Marani M, Prager K, Williams T, Roberts B, Salmon K, Gondo P, Hadebe B, Kayani A, Masunda B, Ahmed A, Morris A, Jakkula S, Long K, Whiteley S, Wilby E, Ogg B, Moultrie S, Odam M, Bewley J, Garland Z, Grimmer L, Gumbrill B, Johnson R, Sweet K, Webster D, Efford G, Bennett S, Goodwin E, Jackson M, Kent A, Tibke C, Woodyatt W, Zaki A, Daniel A, Finn J, Saha R, Staines N, Easthope A, Bremmer P, Allan J, Geary T, Houston G, Meikle A, O’Brien P, Bell D, Boyle R, Douglas K, Glass L, Lee E, Lennon L, Rattray A, Charnock R, McFarland D, Cosgrove D, Attwood B, Parsons P, Carmody S, Oblak M, Popescu M, Thankachen M, Baruah R, Morris S, Ferguson S, Shepherd A, Altabaibeh A, Alvaro A, Gilbert K, Ma L, Mostoles L, Parmar C, Simpson K, Jetha C, Booker L, Pratley A, Cosier T, Millen G, Richardson N, Schumacher N, Weston H, Rand J, Alex B, Bach B, Barclay WS, Bogaert D, Chand M, Cooke GS, Docherty AB, Dunning J, da Silva Filipe A, Fletcher T, Green CA, Harrison EM, Hiscox JA, Ijaz S, Khoo S, Klenerman P, Lim WS, Mentzer AJ, Merson L, Noursadeghi M, Moore SC, Palmarini M, Paxton WA, Pollakis G, Price N, Rambaut A, Robertson DL, Russell CD, Sancho-Shimizu V, Scott JT, de Silva T, Sigfrid L, Solomon T, Sriskandan S, Stuart D, Tedder RS, Thomson EC, Roger Thompson AA, Thwaites RS, Turtle LCW, Gupta RK, Palmieri C, Swann OV, Zambon M, Dumas M-E, Griffin JL, Takats Z, Chechi K, Andrikopoulos P, Osagie A, Olanipekun M, Liggi S, Lewis MR, Correia G dos S, Sands CJ, Takis P, Maslen L, Greenhalf W, Shaw V, McDonald SE, Keating S, Ahmed KA, Armstrong JA, Ashworth M, Asiimwe IG, Bakshi S, Barlow SL, Booth L, Brennan B, Bullock K, Catterall BWA, Clark JJ, Clarke EA, Cooper L, Cox H, Davis C, Dincarslan O, Dunn C, Dyer P, Elliott A, Evans A, Finch L, Fisher LWS, Foster T, Garcia-Dorival I, Greenhalf W, Gunning P, Hartley C, Jensen RL, Jones CB, Jones TR, Khandaker S, King K, Kiy RT, Koukorava C, Lake A, Lant S, Latawiec D, Lavelle-Langham L, Lefteri D, Lett L, Livoti LA, Mancini M, McDonald S, McEvoy L, McLauchlan J, Metelmann S, Miah NS, Middleton J, Mitchell J, Moore SC, Murphy EG, Penrice-Randal R, Pilgrim J, Prince T, Reynolds W, Ridley PM, Sales D, Shaw VE, Shears RK, Small B, Subramaniam KS, Szemiel A, Taggart A, Tanianis-Hughes J, Thomas J, Trochu E, van Tonder L, Wilcock E, Zhang JE, Flaherty L, Maziere N, Cass E, Carracedo AD, Carlucci N, Holmes A, Massey H, Murphy L, Wrobel N, McCafferty S, Morrice K, MacLean A, Armstrong R, Boz C, Brown A, Clark R, Coutts A, Cullum L, Day N, Donnelly L, Duncan E, Fawkes A, Finernan P, Gilchrist T, Golightly A, Hafezi K, Law D, Law R, Law S, Macgillivray L, Maclean A, Mal H, McCafferty S, Mcmaster E, Meikle J, Moore SC, Morrice K, Murphy L, Oosthuyzen W, Paterson T, Stenhouse A, Swets M, Szoor-McElhinney H, Taneski F, Wackett T, Ward M, Weaver J, Wrobel N, Coyle J, Gallagher B, Lidstone-Scott R, Hamilton D, Schon K, Furlong A, Biggs H, Griffiths F, Andrews E, Brickell K, Smyth M, Murphy L, Carson G, Hardwick H, Donohue C. 2021. Mapping the human genetic architecture of COVID-19. Nature 600:472–477.
Gourraud P-A, Khankhanian P, Cereb N, Yang SY, Feolo M, Maiers M, D. Rioux J, Hauser S, Oksenberg J. 2014. HLA Diversity in the 1000 Genomes Dataset. PLoS ONE 9:e97282.
Zhao J, Yang Y, Huang H, Li D, Gu D, Lu X, Zhang Z, Liu L, Liu T, Liu Y, He Y, Sun B, Wei M, Yang G, Wang X, Zhang L, Zhou X, Xing M, Wang PG. 2020. Relationship between the ABO Blood Group and the COVID-19 Susceptibility. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.11.20031096.
Choi B, Choudhary MC, Regan J, Sparks JA, Padera RF, Qiu X, Solomon IH, Kuo H-H, Boucau J, Bowman K, Adhikari UD, Winkler ML, Mueller AA, Hsu TY-T, Desjardins M, Baden LR, Chan BT, Walker BD, Lichterfeld M, Brigl M, Kwon DS, Kanjilal S, Richardson ET, Jonsson AH, Alter G, Barczak AK, Hanage WP, Yu XG, Gaiha GD, Seaman MS, Cernadas M, Li JZ. 2020. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. New England Journal of Medicine 383:2291–2293.
472.
Andreano E, Piccini G, Licastro D, Casalino L, Johnson NV, Paciello I, Monego SD, Pantano E, Manganaro N, Manenti A, Manna R, Casa E, Hyseni I, Benincasa L, Montomoli E, Amaro RE, McLellan JS, Rappuoli R. 2020. SARS-CoV-2 escape <i>in vitro</i> from a highly neutralizing COVID-19 convalescent plasma. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.12.28.424451.
473.
Lauring AS, Hodcroft EB. 2021. Genetic Variants of SARS-CoV-2—What Do They Mean? JAMA https://doi.org/10.1001/jama.2020.27124.
474.
Oude Munnink BB, Sikkema RS, Nieuwenhuijse DF, Molenaar RJ, Munger E, Molenkamp R, van der Spek A, Tolsma P, Rietveld A, Brouwer M, Bouwmeester-Vincken N, Harders F, Hakze-van der Honing R, Wegdam-Blans MCA, Bouwstra RJ, GeurtsvanKessel C, van der Eijk AA, Velkers FC, Smit LAM, Stegeman A, van der Poel WHM, Koopmans MPG. 2021. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 371:172–177.
475.
Davies NG, Barnard RC, Jarvis CI, Kucharski AJ, Munday J, Pearson CAB, Russell TW, Tully DC, Abbott S, Gimma A, Waites W, Wong KL, van Zandvoort K, Eggo RM, Funk S, Jit M, Atkins KE, Edmunds WJ, CMMID COVID-19 Working Group. 2020. Estimated transmissibility and severity of novel SARS-CoV-2 Variant of Concern 202012/01 in England. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.12.24.20248822.
476.
du Plessis L, McCrone JT, Zarebski AE, Hill V, Ruis C, Gutierrez B, Raghwani J, Ashworth J, Colquhoun R, Connor TR, Faria NR, Jackson B, Loman NJ, O’Toole Á, Nicholls SM, Parag KV, Scher E, Vasylyeva TI, Volz EM, Watts A, Bogoch II, Khan K, Aanensen DM, Kraemer MUG, Rambaut A, Pybus OG, COVID-19 Genomics UK (COG-UK) Consortium†. 2021. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science eabf2946.
Washington NL, White S, Barrett KMS, Cirulli ET, Bolze A, Lu JT. 2020. S gene dropout patterns in SARS-CoV-2 tests suggest spread of the H69del/V70del mutation in the US. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.12.24.20248814.
Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, Doolabh D, Pillay S, San EJ, Msomi N, Mlisana K, von Gottberg A, Walaza S, Allam M, Ismail A, Mohale T, Glass AJ, Engelbrecht S, Van Zyl G, Preiser W, Petruccione F, Sigal A, Hardie D, Marais G, Hsiao M, Korsman S, Davies M-A, Tyers L, Mudau I, York D, Maslo C, Goedhals D, Abrahams S, Laguda-Akingba O, Alisoltani-Dehkordi A, Godzik A, Wibmer CK, Sewell BT, Lourenço J, Alcantara LCJ, Pond SLK, Weaver S, Martin D, Lessells RJ, Bhiman JN, Williamson C, de Oliveira T. 2020. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.12.21.20248640.
493.
Wibmer CK, Ayres F, Hermanus T, Madzivhandila M, Kgagudi P, Oosthuysen B, Lambson BE, de Oliveira T, Vermeulen M, van der Berg K, Rossouw T, Boswell M, Ueckermann V, Meiring S, von Gottberg A, Cohen C, Morris L, Bhiman JN, Moore PL. 2021. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat Med 27:622–625.
494.
Cele S, Gazy I, Jackson L, Hwa S-H, Tegally H, Lustig G, Giandhari J, Pillay S, Wilkinson E, Naidoo Y, Karim F, Ganga Y, Khan K, Bernstein M, Balazs AB, Gosnell BI, Hanekom W, Moosa M-YS, Lessells RJ, de Oliveira T, Sigal A. 2021. Escape of SARS-CoV-2 501Y.V2 from neutralization by convalescent plasma. Nature 593:142–146.
Zhang W, Davis BD, Chen SS, Martinez JMS, Plummer JT, Vail E. 2021. Emergence of a novel SARS-CoV-2 strain in Southern California, USA. Cold Spring Harbor Laboratory https://doi.org/10.1101/2021.01.18.21249786.
Greaney AJ, Loes AN, Crawford KHD, Starr TN, Malone KD, Chu HY, Bloom JD. 2021. Comprehensive mapping of mutations to the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human serum antibodies. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.12.31.425021.
Gu H, Chen Q, Yang G, He L, Fan H, Deng Y-Q, Wang Y, Teng Y, Zhao Z, Cui Y, Li Y, Li X-F, Li J, Zhang N-N, Yang X, Chen S, Guo Y, Zhao G, Wang X, Luo D-Y, Wang H, Yang X, Li Y, Han G, He Y, Zhou X, Geng S, Sheng X, Jiang S, Sun S, Qin C-F, Zhou Y. 2020. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science eabc4730.
McCarthy KR, Rennick LJ, Nambulli S, Robinson-McCarthy LR, Bain WG, Haidar G, Duprex WP. 2021. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.11.19.389916.
510.
Kupferschmidt K. 2021. Viral mutations may cause another ‘very, very bad’ COVID-19 wave, scientists warn. Science https://doi.org/10.1126/science.abg4312.
Wu K, Werner AP, Moliva JI, Koch M, Choi A, Stewart-Jones GBE, Bennett H, Boyoglu-Barnum S, Shi W, Graham BS, Carfi A, Corbett KS, Seder RA, Edwards DK. 2021. mRNA-1273 vaccine induces neutralizing antibodies against spike mutants from global SARS-CoV-2 variants. Cold Spring Harbor Laboratory https://doi.org/10.1101/2021.01.25.427948.
516.
Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Moreira ED, Zerbini C, Bailey R, Swanson KA, Roychoudhury S, Koury K, Li P, Kalina WV, Cooper D, Frenck RW, Hammitt LL, Türeci Ö, Nell H, Schaefer A, Ünal S, Tresnan DB, Mather S, Dormitzer PR, Şahin U, Jansen KU, Gruber WC. 2020. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. New England Journal of Medicine 383:2603–2615.
517.
Walsh EE, Frenck RW, Falsey AR, Kitchin N, Absalon J, Gurtman A, Lockhart S, Neuzil K, Mulligan MJ, Bailey R, Swanson KA, Li P, Koury K, Kalina W, Cooper D, Fontes-Garfias C, Shi P-Y, Türeci Ö, Tompkins KR, Lyke KE, Raabe V, Dormitzer PR, Jansen KU, Şahin U, Gruber WC. 2020. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. New England Journal of Medicine 383:2439–2450.
518.
Xie X, Zou J, Fontes-Garfias CR, Xia H, Swanson KA, Cutler M, Cooper D, Menachery VD, Weaver S, Dormitzer PR, Shi P-Y. 2021. Neutralization of N501Y mutant SARS-CoV-2 by BNT162b2 vaccine-elicited sera. Cold Spring Harbor Laboratory https://doi.org/10.1101/2021.01.07.425740.
519.
Wang Z, Schmidt F, Weisblum Y, Muecksch F, Barnes CO, Finkin S, Schaefer-Babajew D, Cipolla M, Gaebler C, Lieberman JA, Oliveira TY, Yang Z, Abernathy ME, Huey-Tubman KE, Hurley A, Turroja M, West KA, Gordon K, Millard KG, Ramos V, Da Silva J, Xu J, Colbert RA, Patel R, Dizon J, Unson-O’Brien C, Shimeliovich I, Gazumyan A, Caskey M, Bjorkman PJ, Casellas R, Hatziioannou T, Bieniasz PD, Nussenzweig MC. 2021. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Cold Spring Harbor Laboratory https://doi.org/10.1101/2021.01.15.426911.
Collier DA, De Marco A, Ferreira IATM, Meng B, Datir RP, Walls AC, Kemp SA, Bassi J, Pinto D, Silacci-Fregni C, Bianchi S, Tortorici MA, Bowen J, Culap K, Jaconi S, Cameroni E, Snell G, Pizzuto MS, Pellanda AF, Garzoni C, Riva A, Baker S, Dougan G, Hess C, Kingston N, Lehner PJ, Lyons PA, Matheson NJ, Owehand WH, Saunders C, Summers C, Thaventhiran JED, Toshner M, Weekes MP, Bucke A, Calder J, Canna L, Domingo J, Elmer A, Fuller S, Harris J, Hewitt S, Kennet J, Jose S, Kourampa J, Meadows A, O’Brien C, Price J, Publico C, Rastall R, Ribeiro C, Rowlands J, Ruffolo V, Tordesillas H, Bullman B, Dunmore BJ, Fawke S, Gräf S, Hodgson J, Huang C, Hunter K, Jones E, Legchenko E, Matara C, Martin J, Mescia F, O’Donnell C, Pointon L, Pond N, Shih J, Sutcliffe R, Tilly T, Treacy C, Tong Z, Wood J, Wylot M, Bergamaschi L, Betancourt A, Bower G, Cossetti C, De Sa A, Epping M, Grenfell R, Hinch A, Huhn O, Jackson S, Jarvis I, Lewis D, Marsden J, Nice F, Okecha G, Omarjee O, Perera M, Richoz N, Romashova V, Yarkoni NS, Sharma R, Stefanucci L, Stephens J, Strezlecki M, Turner L, De Bie EMDD, Bunclark K, Josipovic M, Mackay M, Rossi S, Selvan M, Spencer S, Yong C, Ansaripour A, Michael A, Mwaura L, Patterson C, Polwarth G, Polgarova P, di Stefano G, Fahey C, Michel R, Bong S-H, Coudert JD, Holmes E, Allison J, Butcher H, Caputo D, Clapham-Riley D, Dewhurst E, Furlong A, Graves B, Gray J, Ivers T, Kasanicki M, Le Gresley E, Linger R, Meloy S, Muldoon F, Ovington N, Papadia S, Phelan I, Stark H, Stirrups KE, Townsend P, Walker N, Webster J, Elmer A, Kingston N, Graves B, McCoy LE, Smith KGC, Bradley JR, Temperton N, Ceron-Gutierrez L, Barcenas-Morales G, Robson SC, Loman NJ, Connor TR, Golubchik T, Martinez Nunez RT, Ludden C, Corden S, Johnston I, Bonsall D, Smith CP, Awan AR, Bucca G, Torok ME, Saeed K, Prieto JA, Jackson DK, Hamilton WL, Snell LB, Moore C, Harrison EM, Goncalves S, Fairley DJ, Loose MW, Watkins J, Livett R, Moses S, Amato R, Nicholls S, Bull M, Smith DL, Barrett J, Aanensen DM, Curran MD, Parmar S, Aggarwal D, Shepherd JG, Parker MD, Glaysher S, Bashton M, Underwood AP, Pacchiarini N, Loveson KF, Carabelli AM, Templeton KE, Langford CF, Sillitoe J, de Silva TI, Wang D, Kwiatkowski D, Rambaut A, O’Grady J, Cottrell S, Holden MTG, Thomson EC, Osman H, Andersson M, Chauhan AJ, Hassan-Ibrahim MO, Lawniczak M, Alderton A, Chand M, Constantinidou C, Unnikrishnan M, Darby AC, Hiscox JA, Paterson S, Martincorena I, Robertson DL, Volz EM, Page AJ, Pybus OG, Bassett AR, Ariani CV, Spencer Chapman MH, Li KK, Shah RN, Jesudason NG, Taha Y, McHugh MP, Dewar R, Jahun AS, McMurray C, Pandey S, McKenna JP, Nelson A, Young GR, McCann CM, Elliott S, Lowe H, Temperton B, Roy S, Price A, Rey S, Wyles M, Rooke S, Shaaban S, de Cesare M, Letchford L, Silveira S, Pelosi E, Wilson-Davies E, Hosmillo M, O’Toole Á, Hesketh AR, Stark R, du Plessis L, Ruis C, Adams H, Bourgeois Y, Michell SL, Grammatopoulos D, Edgeworth J, Breuer J, Todd JA, Fraser C, Buck D, John M, Kay GL, Palmer S, Peacock SJ, Heyburn D, Weldon D, Robinson E, McNally A, Muir P, Vipond IB, Boyes J, Sivaprakasam V, Salluja T, Dervisevic S, Meader EJ, Park NR, Oliver K, Jeffries AR, Ott S, da Silva Filipe A, Simpson DA, Williams C, Masoli JAH, Knight BA, Jones CR, Koshy C, Ash A, Casey A, Bosworth A, Ratcliffe L, Xu-McCrae L, Pymont HM, Hutchings S, Berry L, Jones K, Halstead F, Davis T, Holmes C, Iturriza-Gomara M, Lucaci AO, Randell PA, Cox A, Madona P, Harris KA, Brown JR, Mahungu TW, Irish-Tavares D, Haque T, Hart J, Witele E, Fenton ML, Liggett S, Graham C, Swindells E, Collins J, Eltringham G, Campbell S, McClure PC, Clark G, Sloan TJ, Jones C, Lynch J, Warne B, Leonard S, Durham J, Williams T, Haldenby ST, Storey N, Alikhan N-F, Holmes N, Moore C, Carlile M, Perry M, Craine N, Lyons RA, Beckett AH, Goudarzi S, Fearn C, Cook K, Dent H, Paul H, Davies R, Blane B, Girgis ST, Beale MA, Bellis KL, Dorman MJ, Drury E, Kane L, Kay S, McGuigan S, Nelson R, Prestwood L, Rajatileka S, Batra R, Williams RJ, Kristiansen M, Green A, Justice A, Mahanama AIK, Samaraweera B, Hadjirin NF, Quick J, Poplawski R, Kermack LM, Reynolds N, Hall G, Chaudhry Y, Pinckert ML, Georgana I, Moll RJ, Thornton A, Myers R, Stockton J, Williams CA, Yew WC, Trotter AJ, Trebes A, MacIntyre-Cockett G, Birchley A, Adams A, Plimmer A, Gatica-Wilcox B, McKerr C, Hilvers E, Jones H, Asad H, Coombes J, Evans JM, Fina L, Gilbert L, Graham L, Cronin M, Kumziene-Summerhayes S, Taylor S, Jones S, Groves DC, Zhang P, Gallis M, Louka SF, Starinskij I, Jackson C, Gourtovaia M, Tonkin-Hill G, Lewis K, Tovar-Corona JM, James K, Baxter L, Alam MT, Orton RJ, Hughes J, Vattipally S, Ragonnet-Cronin M, Nascimento FF, Jorgensen D, Boyd O, Geidelberg L, Zarebski AE, Raghwani J, Kraemer MUG, Southgate J, Lindsey BB, Freeman TM, Keatley J-P, Singer JB, de Oliveira Martins L, Yeats CA, Abudahab K, Taylor BEW, Menegazzo M, Danesh J, Hogsden W, Eldirdiri S, Kenyon A, Mason J, Robinson TI, Holmes A, Price J, Hartley JA, Curran T, Mather AE, Shankar G, Jones R, Howe R, Morgan S, Wastenge E, Chapman MR, Mookerjee S, Stanley R, Smith W, Peto T, Eyre D, Crook D, Vernet G, Kitchen C, Gulliver H, Merrick I, Guest M, Munn R, Bradley DT, Wyatt T, Beaver C, Foulser L, Palmer S, Churcher CM, Brooks E, Smith KS, Galai K, McManus GM, Bolt F, Coll F, Meadows L, Attwood SW, Davies A, De Lacy E, Downing F, Edwards S, Scarlett GP, Jeremiah S, Smith N, Leek D, Sridhar S, Forrest S, Cormie C, Gill HK, Dias J, Higginson EE, Maes M, Young J, Wantoch M, Jamrozy D, Lo S, Patel M, Hill V, Bewshea CM, Ellard S, Auckland C, Harrison I, Bishop C, Chalker V, Richter A, Beggs A, Best A, Percival B, Mirza J, Megram O, Mayhew M, Crawford L, Ashcroft F, Moles-Garcia E, Cumley N, Hopes R, Asamaphan P, Niebel MO, Gunson RN, Bradley A, Maclean A, Mollett G, Blacow R, Bird P, Helmer T, Fallon K, Tang J, Hale AD, Macfarlane-Smith LR, Harper KL, Carden H, Machin NW, Jackson KA, Ahmad SSY, George RP, Turtle L, O’Toole E, Watts J, Breen C, Cowell A, Alcolea-Medina A, Charalampous T, Patel A, Levett LJ, Heaney J, Rowan A, Taylor GP, Shah D, Atkinson L, Lee JCD, Westhorpe AP, Jannoo R, Lowe HL, Karamani A, Ensell L, Chatterton W, Pusok M, Dadrah A, Symmonds A, Sluga G, Molnar Z, Baker P, Bonner S, Essex S, Barton E, Padgett D, Scott G, Greenaway J, Payne BAI, Burton-Fanning S, Waugh S, Raviprakash V, Sheriff N, Blakey V, Williams L-A, Moore J, Stonehouse S, Smith L, Davidson RK, Bedford L, Coupland L, Wright V, Chappell JG, Tsoleridis T, Ball J, Khakh M, Fleming VM, Lister MM, Howson-Wells HC, Berry L, Boswell T, Joseph A, Willingham I, Duckworth N, Walsh S, Wise E, Moore N, Mori M, Cortes N, Kidd S, Williams R, Gifford L, Bicknell K, Wyllie S, Lloyd A, Impey R, Malone CS, Cogger BJ, Levene N, Monaghan L, Keeley AJ, Partridge DG, Raza M, Evans C, Johnson K, Betteridge E, Farr BW, Goodwin S, Quail MA, Scott C, Shirley L, Thurston SAJ, Rajan D, Bronner IF, Aigrain L, Redshaw NM, Lensing SV, McCarthy S, Makunin A, Balcazar CE, Gallagher MD, Williamson KA, Stanton TD, Michelsen ML, Warwick-Dugdale J, Manley R, Farbos A, Harrison JW, Sambles CM, Studholme DJ, Lackenby A, Mbisa T, Platt S, Miah S, Bibby D, Manso C, Hubb J, Dabrera G, Ramsay M, Bradshaw D, Schaefer U, Groves N, Gallagher E, Lee D, Williams D, Ellaby N, Hartman H, Manesis N, Patel V, Ledesma J, Twohig KA, Allara E, Pearson C, Cheng JKJ, Bridgewater HE, Frost LR, Taylor-Joyce G, Brown PE, Tong L, Broos A, Mair D, Nichols J, Carmichael SN, Smollett KL, Nomikou K, Aranday-Cortes E, Johnson N, Nickbakhsh S, Vamos EE, Hughes M, Rainbow L, Eccles R, Nelson C, Whitehead M, Gregory R, Gemmell M, Wierzbicki C, Webster HJ, Fisher CL, Signell AW, Betancor G, Wilson HD, Nebbia G, Flaviani F, Cerda AC, Merrill TV, Wilson RE, Cotic M, Bayzid N, Thompson T, Acheson E, Rushton S, O’Brien S, Baker DJ, Rudder S, Aydin A, Sang F, Debebe J, Francois S, Vasylyeva TI, Zamudio ME, Gutierrez B, Marchbank A, Maksimovic J, Spellman K, McCluggage K, Morgan M, Beer R, Afifi S, Workman T, Fuller W, Bresner C, Angyal A, Green LR, Parsons PJ, Tucker RM, Brown R, Whiteley M, Bonfield J, Puethe C, Whitwham A, Liddle J, Rowe W, Siveroni I, Le-Viet T, Gaskin A, Johnson R, Abnizova I, Aigrain L, Alderton A, Ali M, Allen L, Amato R, Anderson R, Ariani C, Austin-Guest S, Bala S, Barrett J, Bassett A, Battleday K, Beal J, Beale M, Beaver C, Bellany S, Bellerby T, Bellis K, Berger D, Berriman M, Betteridge E, Bevan P, Binley S, Bishop J, Blackburn K, Bonfield J, Boughton N, Bowker S, Brendler-Spaeth T, Bronner I, Brooklyn T, Buddenborg SK, Bush R, Caetano C, Cagan A, Carter N, Cartwright J, Monteiro TC, Chapman L, Chillingworth T-J, Clapham P, Clark R, Clarke A, Clarke C, Cole D, Cook E, Coppola M, Cornell L, Cornwell C, Corton C, Crackett A, Cranage A, Craven H, Craw S, Crawford M, Cutts T, Dabrowska M, Davies M, Davies R, Dawson J, Day C, Densem A, Dibling T, Dockree C, Dodd D, Dogga S, Dorman M, Dougan G, Dougherty M, Dove A, Drummond L, Drury E, Dudek M, Durham J, Durrant L, Easthope E, Eckert S, Ellis P, Farr B, Fenton M, Ferrero M, Flack N, Fordham H, Forsythe G, Foulser L, Francis M, Fraser A, Freeman A, Galvin A, Garcia-Casado M, Gedny A, Girgis S, Glover J, Goncalves S, Goodwin S, Gould O, Gourtovaia M, Gray A, Gray E, Griffiths C, Gu Y, Guerin F, Hamilton W, Hanks H, Harrison E, Harrott A, Harry E, Harvison J, Heath P, Hernandez-Koutoucheva A, Hobbs R, Holland D, Holmes S, Hornett G, Hough N, Huckle L, Hughes-Hallet L, Hunter A, Inglis S, Iqbal S, Jackson A, Jackson D, James K, Jamrozy D, Verdejo CJ, Johnston I, Jones M, Kallepally K, Kane L, Kay K, Kay S, Keatley J, Keith A, King A, Kitchin L, Kleanthous M, Klimekova M, Korlevic P, Krasheninnkova K, Kwiatkowski D, Lane G, Langford C, Laverack A, Law K, Lawniczak M, Lensing S, Leonard S, Letchford L, Lewis K, Lewis-Wade A, Liddle J, Lin Q, Lindsay S, Linsdell S, Livett R, Lo S, Long R, Lovell J, Lovell J, Ludden C, Mack J, Maddison M, Makunin A, Mamun I, Mansfield J, Marriott N, Martin M, Martincorena I, Mayho M, McCarthy S, McClintock J, McGuigan S, McHugh S, McMinn L, Meadows C, Mobley E, Moll R, Morra M, Morrow L, Murie K, Nash S, Nathwani C, Naydenova P, Neaverson A, Nelson R, Nerou E, Nicholson J, Nimz T, Noell GG, O’Meara S, Ohan V, Oliver K, Olney C, Ormond D, Oszlanczi A, Palmer S, Pang YF, Pardubska B, Park N, Parmar A, Patel G, Patel M, Payne M, Peacock S, Petersen A, Plowman D, Preston T, Prestwood L, Puethe C, Quail M, Rajan D, Rajatileka S, Rance R, Rawlings S, Redshaw N, Reynolds J, Reynolds M, Rice S, Richardson M, Roberts C, Robinson K, Robinson M, Robinson D, Rogers H, Rojo EM, Roopra D, Rose M, Rudd L, Sadri R, Salmon N, Saul D, Schwach F, Scott C, Seekings P, Shirley L, Sillitoe J, Simms A, Sinnott M, Sivadasan S, Siwek B, Sizer D, Skeldon K, Skelton J, Slater-Tunstill J, Sloper L, Smerdon N, Smith C, Smith C, Smith J, Smith K, Smith M, Smith S, Smith T, Sneade L, Soria CD, Sousa C, Souster E, Sparkes A, Spencer-Chapman M, Squares J, Stanley R, Steed C, Stickland T, Still I, Stratton M, Strickland M, Swann A, Swiatkowska A, Sycamore N, Swift E, Symons E, Szluha S, Taluy E, Tao N, Taylor K, Taylor S, Thompson S, Thompson M, Thomson M, Thomson N, Thurston S, Tonkin-Hill G, Toombs D, Topping B, Tovar-Corona J, Ungureanu D, Uphill J, Urbanova J, Van PJ, Vancollie V, Voak P, Walker D, Walker M, Waller M, Ward G, Weatherhogg C, Webb N, Weldon D, Wells A, Wells E, Westwood L, Whipp T, Whiteley T, Whitton G, Whitwham A, Widaa S, Williams M, Wilson M, Wright S, Harvey W, Virgin HW, Lanzavecchia A, Piccoli L, Doffinger R, Wills M, Veesler D, Corti D, Gupta RK. 2021. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 593:136–141.
Liu Y, Liu J, Xia H, Zhang X, Fontes-Garfias CR, Swanson KA, Cai H, Sarkar R, Chen W, Cutler M, Cooper D, Weaver SC, Muik A, Sahin U, Jansen KU, Xie X, Dormitzer PR, Shi P-Y. 2021. Neutralizing Activity of BNT162b2-Elicited Serum. N Engl J Med 384:1466–1468.
Mathieu E, Ritchie H, Rodés-Guirao L, Appel C, Giattino C, Hasell J, Macdonald B, Dattani S, Beltekian D, Ortiz-Ospina E, Roser M. 2020. Coronavirus Pandemic (COVID-19). Our World in Data.
533.
Knyazev S, Chhugani K, Sarwal V, Ayyala R, Singh H, Karthikeyan S, Deshpande D, Baykal PI, Comarova Z, Lu A, Porozov Y, Vasylyeva TI, Wertheim JO, Tierney BT, Chiu CY, Sun R, Wu A, Abedalthagafi MS, Pak VM, Nagaraj SH, Smith AL, Skums P, Pasaniuc B, Komissarov A, Mason CE, Bortz E, Lemey P, Kondrashov F, Beerenwinkel N, Lam TT-Y, Wu NC, Zelikovsky A, Knight R, Crandall KA, Mangul S. 2022. Unlocking capacities of genomics for the COVID-19 response and future pandemics. Nat Methods 19:374–380.
Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DK, Bleicker T, Brünink S, Schneider J, Schmidt ML, Mulders DG, Haagmans BL, van der Veer B, van den Brink S, Wijsman L, Goderski G, Romette J-L, Ellis J, Zambon M, Peiris M, Goossens H, Reusken C, Koopmans MP, Drosten C. 2020. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 25.
557.
Garibyan L, Avashia N. 2013. Polymerase Chain Reaction. Journal of Investigative Dermatology 133:1–4.
van Kampen JJA, van de Vijver DAMC, Fraaij PLA, Haagmans BL, Lamers MM, Okba N, van den Akker JPC, Endeman H, Gommers DAMPJ, Cornelissen JJ, Hoek RAS, van der Eerden MM, Hesselink DA, Metselaar HJ, Verbon A, de Steenwinkel JEM, Aron GI, van Gorp ECM, van Boheemen S, Voermans JC, Boucher CAB, Molenkamp R, Koopmans MPG, Geurtsvankessel C, van der Eijk AA. 2021. Duration and key determinants of infectious virus shedding in hospitalized patients with coronavirus disease-2019 (COVID-19). Nat Commun 12.
Brito AF, Semenova E, Dudas G, Hassler GW, Kalinich CC, Kraemer MUG, Ho J, Tegally H, Githinji G, Agoti CN, Matkin LE, Whittaker C, Howden BP, Sintchenko V, Zuckerman NS, Mor O, Blankenship HM, Oliveira T de, Lin RTP, Siqueira MM, Resende PC, Vasconcelos ATR, Spilki FR, Aguiar RS, Alexiev I, Ivanov IN, Philipova I, Carrington CVF, Sahadeo NSD, Gurry C, Maurer-Stroh S, Naidoo D, von Eije KJ, Perkins MD, Kerkhove M van, Hill SC, Sabino EC, Pybus OG, Dye C, Bhatt S, Flaxman S, Suchard MA, Grubaugh ND, Baele G, Faria NR. 2021. Global disparities in SARS-CoV-2 genomic surveillance. Cold Spring Harbor Laboratory.
586.
Yelin I, Aharony N, Tamar ES, Argoetti A, Messer E, Berenbaum D, Shafran E, Kuzli A, Gandali N, Shkedi O, Hashimshony T, Mandel-Gutfreund Y, Halberthal M, Geffen Y, Szwarcwort-Cohen M, Kishony R. 2020. Evaluation of COVID-19 RT-qPCR Test in Multi sample Pools. Clinical Infectious Diseases 71:2073–2078.
587.
Nelson AC, Auch B, Schomaker M, Gohl DM, Grady P, Johnson D, Kincaid R, Karnuth KE, Daniel J, Fiege JK, Fay EJ, Bold T, Langlois RA, Beckman KB, Yohe S. 2020. Analytical Validation of a COVID-19 qRT-PCR Detection Assay Using a 384-well Format and Three Extraction Methods. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.04.02.022186.
Joachim A, Dewald F, Suárez I, Zemlin M, Lang I, Stutz R, Marthaler A, Bosse HM, Lübke N, Münch J, Bernard M-A, Jeltsch K, Tönshoff B, Weidner N, Kräusslich H-G, Birzele L, Hübner J, Schmied P, Meyer-Bühn M, Horemheb-Rubio G, Cornely OA, Haverkamp H, Wiesmüller G, Fätkenheuer G, Hero B, Kaiser R, Dötsch J, Rybniker J, Cosgun ZC, Hünseler C, Schönenkorb J, Wurm J, Klein F, Heger E, Knops E, Sierra-Aragón S, Kretschmer AC, Sprute R, Kossow A, Hellmich M, Shah-Hosseini K, Weiss M, Goedicke-Fritz S, Kaiser E, Meyer S, Seiwert N, Smola S, Pfuhl T, Lohse S, Schupp A-K, Timm J, Gröne N, Lesmann H, Bredahl R, Schneble L, Turinsky M, Patry C, Hoffmann GF, Müller B, Börner K, Schnitzler P, Heuser A-M, Welker A, von Both U, Kern A. 2021. Pooled RT-qPCR testing for SARS-CoV-2 surveillance in schools - a cluster randomised trial. EClinicalMedicine 39:101082.
Metsky HC, Freije CA, Kosoko-Thoroddsen T-SF, Sabeti PC, Myhrvold C. 2020. CRISPR-based surveillance for COVID-19 using genomically-comprehensive machine learning design. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.26.967026.
Broughton JP, Deng X, Yu G, Fasching CL, Servellita V, Singh J, Miao X, Streithorst JA, Granados A, Sotomayor-Gonzalez A, Zorn K, Gopez A, Hsu E, Gu W, Miller S, Pan C-Y, Guevara H, Wadford DA, Chen JS, Chiu CY. 2020. CRISPR–Cas12-based detection of SARS-CoV-2. Nature Biotechnology 38:870–874.
604.
Lucia C, Federico P-B, Alejandra GC. 2020. An ultrasensitive, rapid, and portable coronavirus SARS-CoV-2 sequence detection method based on CRISPR-Cas12. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.29.971127.
605.
Xiong D, Dai W, Gong J, Li G, Liu N, Wu W, Pan J, Chen C, Jiao Y, Deng H, Ye J, Zhang X, Huang H, Li Q, Xue L, Zhang X, Tang G. 2020. Rapid detection of SARS-CoV-2 with CRISPR-Cas12a. PLoS Biol 18:e3000978.
Guo L, Sun X, Wang X, Liang C, Jiang H, Gao Q, Dai M, Qu B, Fang S, Mao Y, Chen Y, Feng G, Gu Q, Wang RR, Zhou Q, Li W. 2020. SARS-CoV-2 detection with CRISPR diagnostics. Cell Discov 6.
Fozouni P, Son S, Díaz de León Derby M, Knott GJ, Gray CN, D’Ambrosio MV, Zhao C, Switz NA, Kumar GR, Stephens SI, Boehm D, Tsou C-L, Shu J, Bhuiya A, Armstrong M, Harris AR, Chen P-Y, Osterloh JM, Meyer-Franke A, Joehnk B, Walcott K, Sil A, Langelier C, Pollard KS, Crawford ED, Puschnik AS, Phelps M, Kistler A, DeRisi JL, Doudna JA, Fletcher DA, Ott M. 2021. Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell 184:323–333.e9.
Adams ER, Ainsworth M, Anand R, Andersson MI, Auckland K, Baillie JK, Barnes E, Beer S, Bell JI, Berry T, Bibi S, Carroll M, Chinnakannan SK, Clutterbuck E, Cornall RJ, Crook DW, de Silva T, Dejnirattisai W, Dingle KE, Dold C, Espinosa A, Eyre DW, Farmer H, Fernandez Mendoza M, Georgiou D, Hoosdally SJ, Hunter A, Jefferey K, Kelly DF, Klenerman P, Knight J, Knowles C, Kwok AJ, Leuschner U, Levin R, Liu C, López-Camacho C, Martinez J, Matthews PC, McGivern H, Mentzer AJ, Milton J, Mongkolsapaya J, Moore SC, Oliveira MS, Pereira F, Perez E, Peto T, Ploeg RJ, Pollard A, Prince T, Roberts DJ, Rudkin JK, Sanchez V, Screaton GR, Semple MG, Slon-Campos J, Skelly DT, Smith EN, Sobrinodiaz A, Staves J, Stuart DI, Supasa P, Surik T, Thraves H, Tsang P, Turtle L, Walker AS, Wang B, Washington C, Watkins N, Whitehouse J. 2020. Antibody testing for COVID-19: A report from the National COVID Scientific Advisory Panel. Wellcome Open Res 5:139.
Service R. 2020. The standard coronavirus test, if available, works well—but can new diagnostics help in this pandemic? Science https://doi.org/10.1126/science.abb8400.
2001. The distribution and functions of immunoglobulin isotypesImmunobiology: The Immune System in Health and Disease. Garland Science. https://www.ncbi.nlm.nih.gov/books/NBK27162.
Dan JM, Mateus J, Kato Y, Hastie KM, Yu ED, Faliti CE, Grifoni A, Ramirez SI, Haupt S, Frazier A, Nakao C, Rayaprolu V, Rawlings SA, Peters B, Krammer F, Simon V, Saphire EO, Smith DM, Weiskopf D, Sette A, Crotty S. 2021. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science eabf4063.
Sherina N, Piralla A, Du L, Wan H, Kumagai-Braesch M, Andréll J, Braesch-Andersen S, Cassaniti I, Percivalle E, Sarasini A, Bergami F, Di Martino R, Colaneri M, Vecchia M, Sambo M, Zuccaro V, Bruno R, Sachs M, Oggionni T, Meloni F, Abolhassani H, Bertoglio F, Schubert M, Byrne-Steele M, Han J, Hust M, Xue Y, Hammarström L, Baldanti F, Marcotte H, Pan-Hammarström Q. 2021. Persistence of SARS-CoV-2-specific B and T cell responses in convalescent COVID-19 patients 6–8 months after the infection. Med 2:281–295.e4.
665.
Gaebler C, Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Tokuyama M, Cho A, Jankovic M, Schaefer-Babajew D, Oliveira TY, Cipolla M, Viant C, Barnes CO, Bram Y, Breton G, Hägglöf T, Mendoza P, Hurley A, Turroja M, Gordon K, Millard KG, Ramos V, Schmidt F, Weisblum Y, Jha D, Tankelevich M, Martinez-Delgado G, Yee J, Patel R, Dizon J, Unson-O’Brien C, Shimeliovich I, Robbiani DF, Zhao Z, Gazumyan A, Schwartz RE, Hatziioannou T, Bjorkman PJ, Mehandru S, Bieniasz PD, Caskey M, Nussenzweig MC. 2021. Evolution of antibody immunity to SARS-CoV-2. Nature 591:639–644.
666.
Wajnberg A, Amanat F, Firpo A, Altman DR, Bailey MJ, Mansour M, McMahon M, Meade P, Mendu DR, Muellers K, Stadlbauer D, Stone K, Strohmeier S, Simon V, Aberg J, Reich DL, Krammer F, Cordon-Cardo C. 2020. Robust neutralizing antibodies to SARS-CoV-2 infection persist for months. Science 370:1227–1230.
667.
Seow J, Graham C, Merrick B, Acors S, Pickering S, Steel KJA, Hemmings O, O’Byrne A, Kouphou N, Galao RP, Betancor G, Wilson HD, Signell AW, Winstone H, Kerridge C, Huettner I, Jimenez-Guardeño JM, Lista MJ, Temperton N, Snell LB, Bisnauthsing K, Moore A, Green A, Martinez L, Stokes B, Honey J, Izquierdo-Barras A, Arbane G, Patel A, Tan MKI, O’Connell L, O’Hara G, MacMahon E, Douthwaite S, Nebbia G, Batra R, Martinez-Nunez R, Shankar-Hari M, Edgeworth JD, Neil SJD, Malim MH, Doores KJ. 2020. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS-CoV-2 infection in humans. Nat Microbiol 5:1598–1607.
668.
Wheatley AK, Juno JA, Wang JJ, Selva KJ, Reynaldi A, Tan H-X, Lee WS, Wragg KM, Kelly HG, Esterbauer R, Davis SK, Kent HE, Mordant FL, Schlub TE, Gordon DL, Khoury DS, Subbarao K, Cromer D, Gordon TP, Chung AW, Davenport MP, Kent SJ. 2021. Evolution of immune responses to SARS-CoV-2 in mild-moderate COVID-19. Nat Commun 12.
669.
Garcia-Beltran WF, Lam EC, Astudillo MG, Yang D, Miller TE, Feldman J, Hauser BM, Caradonna TM, Clayton KL, Nitido AD, Murali MR, Alter G, Charles RC, Dighe A, Branda JA, Lennerz JK, Lingwood D, Schmidt AG, Iafrate AJ, Balazs AB. 2021. COVID-19-neutralizing antibodies predict disease severity and survival. Cell 184:476–488.e11.
670.
Dowell AC, Butler MS, Jinks E, Tut G, Lancaster T, Sylla P, Begum J, Bruton R, Pearce H, Verma K, Logan N, Tyson G, Spalkova E, Margielewska-Davies S, Taylor GS, Syrimi E, Baawuah F, Beckmann J, Okike IO, Ahmad S, Garstang J, Brent AJ, Brent B, Ireland G, Aiano F, Amin-Chowdhury Z, Jones S, Borrow R, Linley E, Wright J, Azad R, Waiblinger D, Davis C, Thomson EC, Palmarini M, Willett BJ, Barclay WS, Poh J, Amirthalingam G, Brown KE, Ramsay ME, Zuo J, Moss P, Ladhani S. 2021. Children develop robust and sustained cross-reactive spike-specific immune responses to SARS-CoV-2 infection. Nat Immunol 23:40–49.
Kaneko N, Kuo H-H, Boucau J, Farmer JR, Allard-Chamard H, Mahajan VS, Piechocka-Trocha A, Lefteri K, Osborn M, Bals J, Bartsch YC, Bonheur N, Caradonna TM, Chevalier J, Chowdhury F, Diefenbach TJ, Einkauf K, Fallon J, Feldman J, Finn KK, Garcia-Broncano P, Hartana CA, Hauser BM, Jiang C, Kaplonek P, Karpell M, Koscher EC, Lian X, Liu H, Liu J, Ly NL, Michell AR, Rassadkina Y, Seiger K, Sessa L, Shin S, Singh N, Sun W, Sun X, Ticheli HJ, Waring MT, Zhu AL, Alter G, Li JZ, Lingwood D, Schmidt AG, Lichterfeld M, Walker BD, Yu XG, Padera RF, Pillai S. 2020. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell 183:143–157.e13.
673.
Zuo J, Dowell A, Pearce H, Verma K, Long H, Begum J, Aiano F, Amin-Chowdhury Z, Hallis B, Stapley L, Borrow R, Linley E, Ahmad S, Parker B, Horsley A, Amirthalingam G, Brown K, Ramsay M, Ladhani S, Moss P. 2020. Robust SARS-CoV-2-specific T-cell immunity is maintained at 6 months following primary infection. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.11.01.362319.
674.
Breton G, Mendoza P, Hagglof T, Oliveira TY, Schaefer-Babajew D, Gaebler C, Turroja M, Hurley A, Caskey M, Nussenzweig MC. 2020. Persistent Cellular Immunity to SARS-CoV-2 Infection. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.12.08.416636.
Huang AT, Garcia-Carreras B, Hitchings MDT, Yang B, Katzelnick L, Rattigan SM, Borgert B, Moreno C, Solomon BD, Rodriguez-Barraquer I, Lessler J, Salje H, Burke DS, Wesolowski A, Cummings DAT. 2020. A systematic review of antibody mediated immunity to coronaviruses: antibody kinetics, correlates of protection, and association of antibody responses with severity of disease. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.04.14.20065771.
Roberts M, Driggs D, Thorpe M, Gilbey J, Yeung M, Ursprung S, Aviles-Rivero AI, Etmann C, McCague C, Beer L, Weir-McCall JR, Teng Z, Gkrania-Klotsas E, Ruggiero A, Korhonen A, Jefferson E, Ako E, Langs G, Gozaliasl G, Yang G, Prosch H, Preller J, Stanczuk J, Tang J, Hofmanninger J, Babar J, Sánchez LE, Thillai M, Gonzalez PM, Teare P, Zhu X, Patel M, Cafolla C, Azadbakht H, Jacob J, Lowe J, Zhang K, Bradley K, Wassin M, Holzer M, Ji K, Ortet MD, Ai T, Walton N, Lio P, Stranks S, Shadbahr T, Lin W, Zha Y, Niu Z, Rudd JHF, Sala E, Schönlieb C-B. 2021. Common pitfalls and recommendations for using machine learning to detect and prognosticate for COVID-19 using chest radiographs and CT scans. Nat Mach Intell 3:199–217.
Killingley B, Mann AJ, Kalinova M, Boyers A, Goonawardane N, Zhou J, Lindsell K, Hare SS, Brown J, Frise R, Smith E, Hopkins C, Noulin N, Löndt B, Wilkinson T, Harden S, McShane H, Baillet M, Gilbert A, Jacobs M, Charman C, Mande P, Nguyen-Van-Tam JS, Semple MG, Read RC, Ferguson NM, Openshaw PJ, Rapeport G, Barclay WS, Catchpole AP, Chiu C. 2022. Safety, tolerability and viral kinetics during SARS-CoV-2 human challenge in young adults. Nat Med 28:1031–1041.
721.
Lordan R, Prior S, Hennessy E, Naik A, Ghosh S, Paschos GK, Skarke C, Barekat K, Hollingsworth T, Juska S, Mazaleuskaya LL, Teegarden S, Glascock AL, Anderson S, Meng H, Tang S-Y, Weljie A, Bottalico L, Ricciotti E, Cherfane P, Mrcela A, Grant G, Poole K, Mayer N, Waring M, Adang L, Becker J, Fries S, FitzGerald GA, Grosser T. 2021. Considerations for the Safe Operation of Schools During the Coronavirus Pandemic. Front Public Health 9.
Siemieniuk RA, Bartoszko JJ, Zeraatkar D, Kum E, Qasim A, Díaz Martinez JP, Izcovich A, Rochwerg B, Lamontagne F, Han MA, Agarwal A, Agoritsas T, Azab M, Bravo G, Chu DK, Couban R, Cusano E, Devji T, Escamilla Z, Foroutan F, Gao Y, Ge L, Ghadimi M, Heels-Ansdell D, Honarmand K, Hou L, Ibrahim S, Khamis A, Lam B, Mansilla C, Loeb M, Miroshnychenko A, Marcucci M, McLeod SL, Motaghi S, Murthy S, Mustafa RA, Pardo-Hernandez H, Rada G, Rizwan Y, Saadat P, Switzer C, Thabane L, Tomlinson G, Vandvik PO, Vernooij RW, Viteri-García A, Wang Y, Yao L, Zhao Y, Guyatt GH, Brignardello-Petersen R. 2020. Drug treatments for covid-19: living systematic review and network meta-analysis. BMJ m2980.
741.
Elezkurtaj S, Greuel S, Ihlow J, Michaelis E, Bischoff P, Kunze CA, Sinn BV, Gerhold M, Hauptmann K, Ingold-Heppner B, Miller F, Herbst H, Corman VM, Martin H, Radbruch H, Heppner FL, Horst D. 2020. Causes of Death and Comorbidities in Patients with COVID-19. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.06.15.20131540.
Fajgenbaum DC, June CH. 2020. Cytokine Storm. New England Journal of Medicine 383:2255–2273.
745.
Nicholls JM, Poon LL, Lee KC, Ng WF, Lai ST, Leung CY, Chu CM, Hui PK, Mak KL, Lim W, Yan KW, Chan KH, Tsang NC, Guan Y, Yuen KY, Malik Peiris J. 2003. Lung pathology of fatal severe acute respiratory syndrome. The Lancet 361:1773–1778.
Prager R, Pratte MT, Unni RR, Bala S, Ng Fat Hing N, Wu K, McGrath TA, Thomas A, Thoma B, Kyeremanteng K. 2021. Content Analysis and Characterization of Medical Tweets During the Early Covid-19 Pandemic. Cureus https://doi.org/10.7759/cureus.13594.
Furuta Y, Takahashi K, Kuno-Maekawa M, Sangawa H, Uehara S, Kozaki K, Nomura N, Egawa H, Shiraki K. 2005. Mechanism of Action of T-705 against Influenza Virus. Antimicrobial Agents and Chemotherapy 49:981–986.
Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, Hohmann E, Chu HY, Luetkemeyer A, Kline S, Lopez de Castilla D, Finberg RW, Dierberg K, Tapson V, Hsieh L, Patterson TF, Paredes R, Sweeney DA, Short WR, Touloumi G, Lye DC, Ohmagari N, Oh M, Ruiz-Palacios GM, Benfield T, Fätkenheuer G, Kortepeter MG, Atmar RL, Creech CB, Lundgren J, Babiker AG, Pett S, Neaton JD, Burgess TH, Bonnett T, Green M, Makowski M, Osinusi A, Nayak S, Lane HC. 2020. Remdesivir for the Treatment of Covid-19 — Final Report. New England Journal of Medicine 383:1813–1826.
Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A, Feldt T, Green G, Green ML, Lescure F-X, Nicastri E, Oda R, Yo K, Quiros-Roldan E, Studemeister A, Redinski J, Ahmed S, Bernett J, Chelliah D, Chen D, Chihara S, Cohen SH, Cunningham J, D’Arminio Monforte A, Ismail S, Kato H, Lapadula G, L’Her E, Maeno T, Majumder S, Massari M, Mora-Rillo M, Mutoh Y, Nguyen D, Verweij E, Zoufaly A, Osinusi AO, DeZure A, Zhao Y, Zhong L, Chokkalingam A, Elboudwarej E, Telep L, Timbs L, Henne I, Sellers S, Cao H, Tan SK, Winterbourne L, Desai P, Mera R, Gaggar A, Myers RP, Brainard DM, Childs R, Flanigan T. 2020. Compassionate Use of Remdesivir for Patients with Severe Covid-19. New England Journal of Medicine 382:2327–2336.
Mohan A, Tiwari P, Suri T, Mittal S, Patel A, Jain A, T. V, Das UK, Bopanna TK, Pandey R, Shelke S, Singh AR, Bhatnagar S, Masih S, Mahajan S, Dwivedi T, Sahoo B, Pandit A, Bhopale S, Vig S, Gupta R, Madan K, Hadda V, Gupta N, Garg R, Meena VP, Guleria R. 2021. Ivermectin in mild and moderate COVID-19 (RIVET-COV): a randomized, placebo-controlled trial. Research Square Platform LLC.
Cohen JB, Hanff TC, William P, Sweitzer N, Rosado-Santander NR, Medina C, Rodriguez-Mori JE, Renna N, Chang TI, Corrales-Medina V, Andrade-Villanueva JF, Barbagelata A, Cristodulo-Cortez R, Díaz-Cucho OA, Spaak J, Alfonso CE, Valdivia-Vega R, Villavicencio-Carranza M, Ayala-García RJ, Castro-Callirgos CA, González-Hernández LA, Bernales-Salas EF, Coacalla-Guerra JC, Salinas-Herrera CD, Nicolosi L, Basconcel M, Byrd JB, Sharkoski T, Bendezú-Huasasquiche LE, Chittams J, Edmonston DL, Vasquez CR, Chirinos JA. 2021. Continuation versus discontinuation of renin–angiotensin system inhibitors in patients admitted to hospital with COVID-19: a prospective, randomised, open-label trial. The Lancet Respiratory Medicine 9:275–284.
2020. Frequently Asked Questions on the Revocation of the Emergency Use Authorization for Hydroxychloroquine Sulfate and Chloroquine Phosphate. US Food and Drug Administration. https://www.fda.gov/media/138946/download.
Guaraldi G, Meschiari M, Cozzi-Lepri A, Milic J, Tonelli R, Menozzi M, Franceschini E, Cuomo G, Orlando G, Borghi V, Santoro A, Di Gaetano M, Puzzolante C, Carli F, Bedini A, Corradi L, Fantini R, Castaniere I, Tabbì L, Girardis M, Tedeschi S, Giannella M, Bartoletti M, Pascale R, Dolci G, Brugioni L, Pietrangelo A, Cossarizza A, Pea F, Clini E, Salvarani C, Massari M, Viale PL, Mussini C. 2020. Tocilizumab in patients with severe COVID-19: a retrospective cohort study. The Lancet Rheumatology 2:e474–e484.
889.
Price CC, Altice FL, Shyr Y, Koff A, Pischel L, Goshua G, Azar MM, Mcmanus D, Chen S-C, Gleeson SE, Britto CJ, Azmy V, Kaman K, Gaston DC, Davis M, Burrello T, Harris Z, Villanueva MS, Aoun-Barakat L, Kang I, Seropian S, Chupp G, Bucala R, Kaminski N, Lee AI, LoRusso PM, Topal JE, Dela Cruz C, Malinis M. 2020. Tocilizumab Treatment for Cytokine Release Syndrome in Hospitalized Patients With Coronavirus Disease 2019. Chest 158:1397–1408.
Rosas IO, Bräu N, Waters M, Go RC, Hunter BD, Bhagani S, Skiest D, Aziz MS, Cooper N, Douglas IS, Savic S, Youngstein T, Del Sorbo L, Cubillo Gracian A, De La Zerda DJ, Ustianowski A, Bao M, Dimonaco S, Graham E, Matharu B, Spotswood H, Tsai L, Malhotra A. 2021. Tocilizumab in Hospitalized Patients with Severe Covid-19 Pneumonia. N Engl J Med 384:1503–1516.
896.
Salama C, Han J, Yau L, Reiss WG, Kramer B, Neidhart JD, Criner GJ, Kaplan-Lewis E, Baden R, Pandit L, Cameron ML, Garcia-Diaz J, Chávez V, Mekebeb-Reuter M, Lima de Menezes F, Shah R, González-Lara MF, Assman B, Freedman J, Mohan SV. 2021. Tocilizumab in Patients Hospitalized with Covid-19 Pneumonia. N Engl J Med 384:20–30.
Joyner MJ, Carter RE, Senefeld JW, Klassen SA, Mills JR, Johnson PW, Theel ES, Wiggins CC, Bruno KA, Klompas AM, Lesser ER, Kunze KL, Sexton MA, Diaz Soto JC, Baker SE, Shepherd JRA, van Helmond N, Verdun NC, Marks P, van Buskirk CM, Winters JL, Stubbs JR, Rea RF, Hodge DO, Herasevich V, Whelan ER, Clayburn AJ, Larson KF, Ripoll JG, Andersen KJ, Buras MR, Vogt MNP, Dennis JJ, Regimbal RJ, Bauer PR, Blair JE, Paneth NS, Fairweather D, Wright RS, Casadevall A. 2021. Convalescent Plasma Antibody Levels and the Risk of Death from Covid-19. New England Journal of Medicine NEJMoa2031893.
909.
, Horby PW, Estcourt L, Peto L, Emberson JR, Staplin N, Spata E, Pessoa-Amorim G, Campbell M, Roddick A, Brunskill NE, George T, Zehnder D, Tiberi S, Aung NN, Uriel A, Widdrington J, Koshy G, Brown T, Scott S, Baillie JK, Buch MH, Chappell LC, Day JN, Faust SN, Jaki T, Jeffery K, Juszczak E, Lim WS, Montgomery A, Mumford A, Rowan K, Thwaites G, Mafham M, Roberts D, Haynes R, Landray MJ. 2021. Convalescent plasma in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Cold Spring Harbor Laboratory.
Sun C, Chen L, Yang J, Luo C, Zhang Y, Li J, Yang J, Zhang J, Xie L. 2020. SARS-CoV-2 and SARS-CoV Spike-RBD Structure and Receptor Binding Comparison and Potential Implications on Neutralizing Antibody and Vaccine Development. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.16.951723.
Pinto D, Park Y-J, Beltramello M, Walls AC, Tortorici MA, Bianchi S, Jaconi S, Culap K, Zatta F, De Marco A, Peter A, Guarino B, Spreafico R, Cameroni E, Case JB, Chen RE, Havenar-Daughton C, Snell G, Telenti A, Virgin HW, Lanzavecchia A, Diamond MS, Fink K, Veesler D, Corti D. 2020. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 583:290–295.
916.
Wang C, Li W, Drabek D, Okba NMA, van Haperen R, Osterhaus ADME, van Kuppeveld FJM, Haagmans BL, Grosveld F, Bosch B-J. 2020. A human monoclonal antibody blocking SARS-CoV-2 infection. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.11.987958.
Hansen J, Baum A, Pascal KE, Russo V, Giordano S, Wloga E, Fulton BO, Yan Y, Koon K, Patel K, Chung KM, Hermann A, Ullman E, Cruz J, Rafique A, Huang T, Fairhurst J, Libertiny C, Malbec M, Lee W, Welsh R, Farr G, Pennington S, Deshpande D, Cheng J, Watty A, Bouffard P, Babb R, Levenkova N, Chen C, Zhang B, Romero Hernandez A, Saotome K, Zhou Y, Franklin M, Sivapalasingam S, Lye DC, Weston S, Logue J, Haupt R, Frieman M, Chen G, Olson W, Murphy AJ, Stahl N, Yancopoulos GD, Kyratsous CA. 2020. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 369:1010–1014.
Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, Musser BJ, Soo Y, Rofail D, Im J, Perry C, Pan C, Hosain R, Mahmood A, Davis JD, Turner KC, Hooper AT, Hamilton JD, Baum A, Kyratsous CA, Kim Y, Cook A, Kampman W, Kohli A, Sachdeva Y, Graber X, Kowal B, DiCioccio T, Stahl N, Lipsich L, Braunstein N, Herman G, Yancopoulos GD. 2021. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with Covid-19. N Engl J Med 384:238–251.
Gupta A, Gonzalez-Rojas Y, Juarez E, Casal MC, Moya J, Falci DR, Sarkis E, Solis J, Zheng H, Scott N, Cathcart AL, Hebner CM, Sager J, Mogalian E, Tipple C, Peppercorn A, Alexander E, Pang PS, Free A, Brinson C, Aldinger M, Shapiro AE. 2021. Early Covid-19 Treatment With SARS-CoV-2 Neutralizing Antibody Sotrovimab. Cold Spring Harbor Laboratory.
Diamond M, Chen R, Xie X, Case J, Zhang X, VanBlargan L, Liu Y, Liu J, Errico J, Winkler E, Suryadevara N, Tahan S, Turner J, Kim W, Schmitz A, Thapa M, Wang D, Boon A, Pinto D, Presti R, O’Halloran J, Kim A, Deepak P, Fremont D, Corti D, Virgin H, Crowe J, Droit L, Ellebedy A, Shi P-Y, Gilchuk P. 2021. SARS-CoV-2 variants show resistance to neutralization by many monoclonal and serum-derived polyclonal antibodies. Research Square Platform LLC.
Wang P, Nair MS, Liu L, Iketani S, Luo Y, Guo Y, Wang M, Yu J, Zhang B, Kwong PD, Graham BS, Mascola JR, Chang JY, Yin MT, Sobieszczyk M, Kyratsous CA, Shapiro L, Sheng Z, Huang Y, Ho DD. 2021. Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7. Cold Spring Harbor Laboratory.
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang X, Bai F, Liu H, Liu X, Guddat LW, Xu W, Xiao G, Qin C, Shi Z, Jiang H, Rao Z, Yang H. 2020. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582:289–293.
Kuleshov MV, Stein DJ, Clarke DJB, Kropiwnicki E, Jagodnik KM, Bartal A, Evangelista JE, Hom J, Cheng M, Bailey A, Zhou A, Ferguson LB, Lachmann A, Ma'ayan A. 2020. The COVID-19 Drug and Gene Set Library. Patterns 1:100090.
976.
Sadegh S, Matschinske J, Blumenthal DB, Galindez G, Kacprowski T, List M, Nasirigerdeh R, Oubounyt M, Pichlmair A, Rose TD, Salgado-Albarrán M, Späth J, Stukalov A, Wenke NK, Yuan K, Pauling JK, Baumbach J. 2020. Exploring the SARS-CoV-2 virus-host-drug interactome for drug repurposing. Nat Commun 11.
Mulangu S, Dodd LE, Davey RT, Tshiani Mbaya O, Proschan M, Mukadi D, Lusakibanza Manzo M, Nzolo D, Tshomba Oloma A, Ibanda A, Ali R, Coulibaly S, Levine AC, Grais R, Diaz J, Lane HC, Muyembe-Tamfum J-J, the PALM Writing Group. 2019. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. New England Journal of Medicine 381:2293–2303.
1017.
Sheahan TP, Sims AC, Graham RL, Menachery VD, Gralinski LE, Case JB, Leist SR, Pyrc K, Feng JY, Trantcheva I, Bannister R, Park Y, Babusis D, Clarke MO, Mackman RL, Spahn JE, Palmiotti CA, Siegel D, Ray AS, Cihlar T, Jordan R, Denison MR, Baric RS. 2017. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Science Translational Medicine 9:eaal3653.
1018.
Cohen J. 2020. Did an experimental drug help a U.S. coronavirus patient? Science https://doi.org/10.1126/science.abb7243.
1019.
Kujawski SA, Wong KK, Collins JP, Epstein L, Killerby ME, Midgley CM, Abedi GR, Ahmed NS, Almendares O, Alvarez FN, Anderson KN, Balter S, Barry V, Bartlett K, Beer K, Ben-Aderet MA, Benowitz I, Biggs H, Binder AM, Black SR, Bonin B, Brown CM, Bruce H, Bryant-Genevier J, Budd A, Buell D, Bystritsky R, Cates J, Charles EM, Chatham-Stephens K, Chea N, Chiou H, Christiansen D, Chu V, Cody S, Cohen M, Conners E, Curns A, Dasari V, Dawson P, DeSalvo T, Diaz G, Donahue M, Donovan S, Duca LM, Erickson K, Esona MD, Evans S, Falk J, Feldstein LR, Fenstersheib M, Fischer M, Fisher R, Foo C, Fricchione MJ, Friedman O, Fry AM, Galang RR, Garcia MM, Gerber SI, Gerrard G, Ghinai I, Gounder P, Grein J, Grigg C, Gunzenhauser JD, Gutkin GI, Haddix M, Hall AJ, Han G, Harcourt J, Harriman K, Haupt T, Haynes A, Holshue M, Hoover C, Hunter JC, Jacobs MW, Jarashow C, Jhung MA, Joshi K, Kamali T, Kamili S, Kim L, Kim M, King J, Kirking HL, Kita-Yarbro A, Klos R, Kobayashi M, Kocharian A, Komatsu KK, Koppaka R, Layden JE, Li Y, Lindquist S, Lindstrom S, Link-Gelles R, Lively J, Livingston M, Lo K, Lo J, Lu X, Lynch B, Madoff L, Malapati L, Marks G, Marlow M, Mathisen GE, McClung N, McGovern O, McPherson TD, Mehta M, Meier A, Mello L, Moon S, Morgan M, Moro RN, Murray J, Murthy R, Novosad S, Oliver SE, O'Shea J, Pacilli M, Paden CR, Pallansch MA, Patel M, Patel S, Pedraza I, Pillai SK, Pindyck T, Pray I, Queen K, Quick N, Reese H, Rha B, Rhodes H, Robinson S, Robinson P, Rolfes M, Routh J, Rubin R, Rudman SL, Sakthivel SK, Scott S, Shepherd C, Shetty V, Smith EA, Smith S, Stierman B, Stoecker W, Sunenshine R, Sy-Santos R, Tamin A, Tao Y, Terashita D, Thornburg NJ, Tong S, Traub E, Tural A, Uehara A, Uyeki TM, Vahey G, Verani JR, Villarino E, Wallace M, Wang L, Watson JT, Westercamp M, Whitaker B, Wilkerson S, Woodruff RC, Wortham JM, Wu T, Xie A, Yousaf A, Zahn M, Zhang J, The COVID-19 Investigation Team. 2020. First 12 patients with coronavirus disease 2019 (COVID-19) in the United States. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.09.20032896.
1020.
Holshue ML, DeBolt C, Lindquist S, Lofy KH, Wiesman J, Bruce H, Spitters C, Ericson K, Wilkerson S, Tural A, Diaz G, Cohn A, Fox L, Patel A, Gerber SI, Kim L, Tong S, Lu X, Lindstrom S, Pallansch MA, Weldon WC, Biggs HM, Uyeki TM, Pillai SK. 2020. First Case of 2019 Novel Coronavirus in the United States. New England Journal of Medicine 382:929–936.
1021.
Goldman JD, Lye DCB, Hui DS, Marks KM, Bruno R, Montejano R, Spinner CD, Galli M, Ahn M-Y, Nahass RG, Chen Y-S, SenGupta D, Hyland RH, Osinusi AO, Cao H, Blair C, Wei X, Gaggar A, Brainard DM, Towner WJ, Muñoz J, Mullane KM, Marty FM, Tashima KT, Diaz G, Subramanian A. 2020. Remdesivir for 5 or 10 Days in Patients with Severe Covid-19. New England Journal of Medicine 383:1827–1837.
Spinner CD, Gottlieb RL, Criner GJ, Arribas López JR, Cattelan AM, Soriano Viladomiu A, Ogbuagu O, Malhotra P, Mullane KM, Castagna A, Chai LYA, Roestenberg M, Tsang OTY, Bernasconi E, Le Turnier P, Chang S-C, SenGupta D, Hyland RH, Osinusi AO, Cao H, Blair C, Wang H, Gaggar A, Brainard DM, McPhail MJ, Bhagani S, Ahn MY, Sanyal AJ, Huhn G, Marty FM, for the GS-US-540-5774 Investigators. 2020. Effect of Remdesivir vs Standard Care on Clinical Status at 11 Days in Patients With Moderate COVID-19. JAMA 324:1048.
1032.
Kalil AC, Patterson TF, Mehta AK, Tomashek KM, Wolfe CR, Ghazaryan V, Marconi VC, Ruiz-Palacios GM, Hsieh L, Kline S, Tapson V, Iovine NM, Jain MK, Sweeney DA, El Sahly HM, Branche AR, Regalado Pineda J, Lye DC, Sandkovsky U, Luetkemeyer AF, Cohen SH, Finberg RW, Jackson PEH, Taiwo B, Paules CI, Arguinchona H, Erdmann N, Ahuja N, Frank M, Oh M, Kim E-S, Tan SY, Mularski RA, Nielsen H, Ponce PO, Taylor BS, Larson L, Rouphael NG, Saklawi Y, Cantos VD, Ko ER, Engemann JJ, Amin AN, Watanabe M, Billings J, Elie M-C, Davey RT, Burgess TH, Ferreira J, Green M, Makowski M, Cardoso A, de Bono S, Bonnett T, Proschan M, Deye GA, Dempsey W, Nayak SU, Dodd LE, Beigel JH. 2020. Baricitinib plus Remdesivir for Hospitalized Adults with Covid-19. New England Journal of Medicine NEJMoa2031994.
1033.
Hinton DM. 2020. Letter of Authorization: EUA for baricitinib (Olumiant), in combination with remdesivir (Veklury), for the treatment of suspected or laboratory confirmed coronavirus disease 2019 (COVID-19). Food and Drug Administration. https://www.fda.gov/media/143822/download.
Chen Z, Hu J, Zhang Z, Jiang S, Han S, Yan D, Zhuang R, Hu B, Zhang Z. 2020. Efficacy of hydroxychloroquine in patients with COVID-19: results of a randomized clinical trial. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.22.20040758.
C.E.Lane J, Weaver J, Kostka K, Duarte-Salles T, Abrahao MTF, Alghoul H, Alser O, Alshammari TM, Biedermann P, Burn E, Casajust P, Conover M, Culhane AC, Davydov A, DuVall SL, Dymshyts D, Fernandez-Bertolin S, Fišter K, Hardin J, Hester L, Hripcsak G, Kent S, Khosla S, Kolovos S, Lambert CG, van der Lei J, Londhe AA, Lynch KE, Makadia R, Margulis AV, Matheny ME, Mehta P, Morales DR, Morgan-Stewart H, Mosseveld M, Newby D, Nyberg F, Ostropolets A, Park RW, Prats-Uribe A, Rao GA, Reich C, Reps J, Rijnbeek P, Kumaran Sathappan SM, Schuemie M, Seager S, Sena A, Shoaibi A, Spotnitz M, Suchard MA, Swerdel J, Torre CO, Vizcaya D, Wen H, de Wilde M, You SC, Zhang L, Zhuk O, Ryan P, Prieto-Alhambra D. 2020. Safety of hydroxychloroquine, alone and in combination with azithromycin, in light of rapid wide-spread use for COVID-19: a multinational, network cohort and self-controlled case series study. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.04.08.20054551.
1054.
Silva Borba MG, Almeida Val FF, Sampaio VS, Araújo Alexandre MA, Melo GC, Brito M, Gomes Mourão MP, Brito-Sousa JD, Baía-da-Silva D, Farias Guerra MV, Abrahão Hajjar L, Pinto RC, Silva Balieiro AA, Naveca FG, Simão Xavier M, Salomão A, Siqueira AM, Schwarzbolt A, Rosa Croda JH, Nogueira ML, Sierra Romero GA, Bassat Q, Fontes CJ, Cláudio Albuquerque B, Daniel-Ribeiro CT, Monteiro WM, Guimarães Lacerda MV, CloroCovid-19 Team. 2020. Chloroquine diphosphate in two different dosages as adjunctive therapy of hospitalized patients with severe respiratory syndrome in the context of coronavirus (SARS-CoV-2) infection: Preliminary safety results of a randomized, double-blinded, phase IIb clinical trial (CloroCovid-19 Study). Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.04.07.20056424.
Tang W, Cao Z, Han M, Wang Z, Chen J, Sun W, Wu Y, Xiao W, Liu S, Chen E, Chen W, Wang X, Yang J, Lin J, Zhao Q, Yan Y, Xie Z, Li D, Yang Y, Liu L, Qu J, Ning G, Shi G, Xie Q. 2020. Hydroxychloroquine in patients mainly with mild to moderate COVID–19: an open–label, randomized, controlled trial. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.04.10.20060558.
1058.
Magagnoli J, Narendran S, Pereira F, Cummings T, Hardin JW, Sutton SS, Ambati J. 2020. Outcomes of hydroxychloroquine usage in United States veterans hospitalized with Covid-19. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.04.16.20065920.
1059.
Mitjà O, Corbacho-Monné M, Ubals M, Tebé C, Peñafiel J, Tobias A, Ballana E, Alemany A, Riera-Martí N, Pérez CA, Suñer C, Laporte P, Admella P, Mitjà J, Clua M, Bertran L, Sarquella M, Gavilán S, Ara J, Argimon JM, Casabona J, Cuatrecasas G, Cañadas P, Elizalde-Torrent A, Fabregat R, Farré M, Forcada A, Flores-Mateo G, Muntada E, Nadal N, Narejos S, Nieto A, Prat N, Puig J, Quiñones C, Reyes-Ureña J, Ramírez-Viaplana F, Ruiz L, Riveira-Muñoz E, Sierra A, Velasco C, Vivanco-Hidalgo RM, Sentís A, G-Beiras C, Clotet B, Vall-Mayans M. 2020. Hydroxychloroquine for Early Treatment of Adults With Mild Coronavirus Disease 2019: A Randomized, Controlled Trial. Clinical Infectious Diseases ciaa1009.
1060.
Boulware DR, Pullen MF, Bangdiwala AS, Pastick KA, Lofgren SM, Okafor EC, Skipper CP, Nascene AA, Nicol MR, Abassi M, Engen NW, Cheng MP, LaBar D, Lother SA, MacKenzie LJ, Drobot G, Marten N, Zarychanski R, Kelly LE, Schwartz IS, McDonald EG, Rajasingham R, Lee TC, Hullsiek KH. 2020. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. New England Journal of Medicine 383:517–525.
Elimination or Prolongation of ACE Inhibitors and ARB in Coronavirus Disease 2019 - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04338009. Retrieved 8 February 2021.
Sparks MA, South A, Welling P, Luther JM, Cohen J, Byrd JB, Burrell LM, Batlle D, Tomlinson L, Bhalla V, Rheault MN, Soler MJ, Swaminathan S, Hiremath S. 2020. Sound Science before Quick Judgement Regarding RAS Blockade in COVID-19. Clinical Journal of the American Society of Nephrology 15:714–716.
1072.
Sparks M, Hiremath S. The Coronavirus Conundrum: ACE2 and Hypertension Edition. NephJC. http://www.nephjc.com/news/covidace2. Retrieved 30 July 2020.
Watad A, Bragazzi NL, Bridgewood C, Mansour M, Mahroum N, Riccò M, Nasr A, Hussein A, Gendelman O, Shoenfeld Y, Lindar M, Amital H, Wu J, McGonagle D. 2020. Systematic Review and Meta-Analysis of Case-Control Studies from 7,000 COVID-19 Pneumonia Patients Suggests a Beneficial Impact of Tocilizumab with Benefit Most Evident in Non-Corticosteroid Exposed Subjects. SSRN Electronic Journal https://doi.org/10.2139/ssrn.3642653.
Kaye A, Siegel R. 2020. The Efficacy of IL-6 Inhibitor Tocilizumab in Reducing Severe COVID-19 Mortality: A Systematic Review. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.07.10.20150938.
2015. Tocilizumab (Actemra): Adult Patients with Moderately to Severely Active Rheumatoid Arthritis. Canadian Agency for Drugs and Technologies in Health, Ottawa (ON). https://www.ncbi.nlm.nih.gov/books/NBK349513/table/T43/.
Mathieu E, Ritchie H, Ortiz-Ospina E, Roser M, Hasell J, Appel C, Giattino C, Rodés-Guirao L. 2021. A global database of COVID-19 vaccinations. Nat Hum Behav 5:947–953.
Jackson LA, Anderson EJ, Rouphael NG, Roberts PC, Makhene M, Coler RN, McCullough MP, Chappell JD, Denison MR, Stevens LJ, Pruijssers AJ, McDermott A, Flach B, Doria-Rose NA, Corbett KS, Morabito KM, O’Dell S, Schmidt SD, Swanson PA, Padilla M, Mascola JR, Neuzil KM, Bennett H, Sun W, Peters E, Makowski M, Albert J, Cross K, Buchanan W, Pikaart-Tautges R, Ledgerwood JE, Graham BS, Beigel JH. 2020. An mRNA Vaccine against SARS-CoV-2 — Preliminary Report. New England Journal of Medicine 383:1920–1931.
Rando HM, Greene CS, Robson MP, Boca SM, Wellhausen N, Lordan R, Brueffer C, Ray S, McGowan LD, Gitter A, Dattoli AA, Velazquez R, Barton JP, Field JM, Ramsundar B, MacLean AL, Lee AJ, Immunology Institute of the Icahn School of Medicine, Hu F, Jadavji NM, Sell E, Wang J, Rafizadeh DN, Skelly AN, Guebila MB, Kolla L, Manheim D, Ghosh S, Byrd JB, Park Y, Bansal V, Capone S, Dziak JJ, Sun Y, Qi Y, Shinholster L, Lukan T, Knyazev S, Perrin D, Mangul S, Das S, Szeto GL, Lubiana T, Mai D, COVID-19 Review Consortium, Goel RR, Boerckel JD, Naik A, Sun Y, Himmelstein DS, Kamil JP, Meyer JG, Mundo AI. 2023. SARS-CoV-2 and COVID-19: An Evolving Review of Diagnostics and Therapeutics.
Jordahl K, Van Den Bossche J, Fleischmann M, McBride J, Wasserman J, Badaracco AG, Gerard J, Snow AD, Tratner J, Perry M, Farmer C, Hjelle GA, Cochran M, Gillies S, Culbertson L, Bartos M, Ward B, Caria G, Taves M, Eubank N, Sangarshanan, Flavin J, Richards M, Rey S, Maxalbert, Bilogur A, Ren C, Arribas-Bel D, Mesejo-León D, Wasser L. 2021. geopandas/geopandas: v0.10.2 (v0.10.2). Zenodo. https://doi.org/gqkzpv.
Gao Q, Bao L, Mao H, Wang L, Xu K, Yang M, Li Y, Zhu L, Wang N, Lv Z, Gao H, Ge X, Kan B, Hu Y, Liu J, Cai F, Jiang D, Yin Y, Qin C, Li J, Gong X, Lou X, Shi W, Wu D, Zhang H, Zhu L, Deng W, Li Y, Lu J, Li C, Wang X, Yin W, Zhang Y, Qin C. 2020. Development of an inactivated vaccine candidate for SARS-CoV-2. Science 369:77–81.
Palacios R, Batista AP, Albuquerque CSN, Patiño EG, Santos J do P, Tilli Reis Pessoa Conde M, Piorelli R de O, Pereira Júnior LC, Raboni SM, Ramos F, Sierra Romero GA, Leal FE, Camargo LFA, Aoki FH, Coelho EB, Oliveira DS, Fontes CJF, Pileggi GCS, Oliveira ALL de, Siqueira AM de, Oliveira DBL de, Botosso VF, Zeng G, Xin Q, Teixeira MM, Nogueira ML, Kallas EG. 2021. Efficacy and Safety of a COVID-19 Inactivated Vaccine in Healthcare Professionals in Brazil: The PROFISCOV Study. SSRN Journal https://doi.org/10.2139/ssrn.3822780.
Jara A, Undurraga EA, González C, Paredes F, Fontecilla T, Jara G, Pizarro A, Acevedo J, Leo K, Leon F, Sans C, Leighton P, Suárez P, García-Escorza H, Araos R. 2021. Effectiveness of an Inactivated SARS-CoV-2 Vaccine in Chile. N Engl J Med 385:875–884.
Xia S, Duan K, Zhang Y, Zhao D, Zhang H, Xie Z, Li X, Peng C, Zhang Y, Zhang W, Yang Y, Chen W, Gao X, You W, Wang X, Wang Z, Shi Z, Wang Y, Yang X, Zhang L, Huang L, Wang Q, Lu J, Yang Y, Guo J, Zhou W, Wan X, Wu C, Wang W, Huang S, Du J, Meng Z, Pan A, Yuan Z, Shen S, Guo W, Yang X. 2020. Effect of an Inactivated Vaccine Against SARS-CoV-2 on Safety and Immunogenicity Outcomes. JAMA 324:951.
1205.
Al Kaabi N, Zhang Y, Xia S, Yang Y, Al Qahtani MM, Abdulrazzaq N, Al Nusair M, Hassany M, Jawad JS, Abdalla J, Hussein SE, Al Mazrouei SK, Al Karam M, Li X, Yang X, Wang W, Lai B, Chen W, Huang S, Wang Q, Yang T, Liu Y, Ma R, Hussain ZM, Khan T, Saifuddin Fasihuddin M, You W, Xie Z, Zhao Y, Jiang Z, Zhao G, Zhang Y, Mahmoud S, ElTantawy I, Xiao P, Koshy A, Zaher WA, Wang H, Duan K, Pan A, Yang X. 2021. Effect of 2 Inactivated SARS-CoV-2 Vaccines on Symptomatic COVID-19 Infection in Adults. JAMA https://doi.org/10.1001/jama.2021.8565.
Jeewandara C, Aberathna IS, Pushpakumara PD, Kamaladasa A, Guruge D, Jayathilaka D, Gunasekara B, Tanussiya S, Kuruppu H, Ranasinghe T, Dayarathne S, Dissanayake O, Gamalath N, Ekanayake D, Jayamali M, Wijesinghe A, Dissanayake M, Madusanka D, Jayadas TT, Mudunkotuwa A, Somathilake G, Harvie M, Nimasha T, Danasekara S, Wijayamuni R, Schimanski L, Tan TK, Dong T, Townsend A, Ogg GS, Malavige GN. 2021. Antibody and T cell responses to Sinopharm/BBIBP-CorV in naïve and previously infected individuals in Sri Lanka. Cold Spring Harbor Laboratory.
Omma A, Batirel A, Aydin M, Yilmaz Karadag F, Erden A, Kucuksahin O, Armagan B, Güven SC, Karakas O, Gokdemir S, Altunal LN, Buber AA, Gemcioglu E, Zengin O, Inan O, Sahiner ES, Korukluoglu G, Sezer Z, Ozdarendeli A, Kara A, Ates I. 2022. Safety and immunogenicity of inactive vaccines as booster doses for COVID-19 in Türkiye: A randomized trial. Human Vaccines & Immunotherapeutics https://doi.org/10.1080/21645515.2022.2122503.
Keech C, Albert G, Cho I, Robertson A, Reed P, Neal S, Plested JS, Zhu M, Cloney-Clark S, Zhou H, Smith G, Patel N, Frieman MB, Haupt RE, Logue J, McGrath M, Weston S, Piedra PA, Desai C, Callahan K, Lewis M, Price-Abbott P, Formica N, Shinde V, Fries L, Lickliter JD, Griffin P, Wilkinson B, Glenn GM. 2020. Phase 1–2 Trial of a SARS-CoV-2 Recombinant Spike Protein Nanoparticle Vaccine. N Engl J Med 383:2320–2332.
1297.
Tian J-H, Patel N, Haupt R, Zhou H, Weston S, Hammond H, Logue J, Portnoff AD, Norton J, Guebre-Xabier M, Zhou B, Jacobson K, Maciejewski S, Khatoon R, Wisniewska M, Moffitt W, Kluepfel-Stahl S, Ekechukwu B, Papin J, Boddapati S, Jason Wong C, Piedra PA, Frieman MB, Massare MJ, Fries L, Bengtsson KL, Stertman L, Ellingsworth L, Glenn G, Smith G. 2021. SARS-CoV-2 spike glycoprotein vaccine candidate NVX-CoV2373 immunogenicity in baboons and protection in mice. Nat Commun 12.
Heath PT, Galiza EP, Baxter DN, Boffito M, Browne D, Burns F, Chadwick DR, Clark R, Cosgrove C, Galloway J, Goodman AL, Heer A, Higham A, Iyengar S, Jamal A, Jeanes C, Kalra PA, Kyriakidou C, McAuley DF, Meyrick A, Minassian AM, Minton J, Moore P, Munsoor I, Nicholls H, Osanlou O, Packham J, Pretswell CH, San Francisco Ramos A, Saralaya D, Sheridan RP, Smith R, Soiza RL, Swift PA, Thomson EC, Turner J, Viljoen ME, Albert G, Cho I, Dubovsky F, Glenn G, Rivers J, Robertson A, Smith K, Toback S. 2021. Safety and Efficacy of NVX-CoV2373 Covid-19 Vaccine. N Engl J Med 385:1172–1183.
1301.
Dunkle LM, Kotloff KL, Gay CL, Áñez G, Adelglass JM, Barrat Hernández AQ, Harper WL, Duncanson DM, McArthur MA, Florescu DF, McClelland RS, Garcia-Fragoso V, Riesenberg RA, Musante DB, Fried DL, Safirstein BE, McKenzie M, Jeanfreau RJ, Kingsley JK, Henderson JA, Lane DC, Ruíz-Palacios GM, Corey L, Neuzil KM, Coombs RW, Greninger AL, Hutter J, Ake JA, Smith K, Woo W, Cho I, Glenn GM, Dubovsky F. 2022. Efficacy and Safety of NVX-CoV2373 in Adults in the United States and Mexico. N Engl J Med 386:531–543.
1302.
Dunkle LM, Kotloff KL, Gay CL, Áñez G, Adelglass JM, Barrat Hernández AQ, Harper WL, Duncanson DM, McArthur MA, Florescu DF, McClelland RS, Garcia-Fragoso V, Riesenberg RA, Musante DB, Fried DL, Safirstein BE, McKenzie M, Jeanfreau RJ, Kingsley JK, Henderson JA, Lane DC, Ruíz-Palacios GM, Corey L, Neuzil KM, Coombs RW, Greninger AL, Hutter J, Ake JA, Smith K, Woo W, Cho I, Glenn GM, Dubovsky F. 2021. Efficacy and Safety of NVX-CoV2373 in Adults in the United States and Mexico. Cold Spring Harbor Laboratory.
Shinde V, Bhikha S, Hoosain Z, Archary M, Bhorat Q, Fairlie L, Lalloo U, Masilela MSL, Moodley D, Hanley S, Fouche L, Louw C, Tameris M, Singh N, Goga A, Dheda K, Grobbelaar C, Kruger G, Carrim-Ganey N, Baillie V, de Oliveira T, Lombard Koen A, Lombaard JJ, Mngqibisa R, Bhorat AE, Benadé G, Lalloo N, Pitsi A, Vollgraaff P-L, Luabeya A, Esmail A, Petrick FG, Oommen-Jose A, Foulkes S, Ahmed K, Thombrayil A, Fries L, Cloney-Clark S, Zhu M, Bennett C, Albert G, Faust E, Plested JS, Robertson A, Neal S, Cho I, Glenn GM, Dubovsky F, Madhi SA. 2021. Efficacy of NVX-CoV2373 Covid-19 Vaccine against the B.1.351 Variant. N Engl J Med 384:1899–1909.
Toledo-Romani ME, García-Carmenate M, Verdecia-Sánchez L, Pérez-Rodríguez S, Rodriguez-González M, Valenzuela-Silva C, Paredes-Moreno B, Sanchez-Ramirez B, González-Mugica R, Hernández-Garcia T, Orosa-Vázquez I, Díaz-Hernández M, Pérez-Guevara MT, Enriquez-Puertas J, Noa-Romero E, Palenzuela-Diaz A, Baro-Roman G, Mendoza-Hernández I, Muñoz Y, Gómez-Maceo Y, Santos-Vega BL, Fernandez-Castillo S, Climent-Ruiz Y, Rodríguez-Noda L, Santana-Mederos D, García-Vega Y, Chen G-W, Doroud D, Biglari A, Boggiano-Ayo T, Valdés-Balbín Y, Rivera DG, García-Rivera D, Vérez-Bencomo V, Cubas-Curbelo M, Rodríguez-Castillo PG, Acevedo-Martínez Y, Estoque-Cabrera S, Ávila-Cabreja JA, Alfaro-Guzmán A, Zulueta-Pérez L, Espino-Rojas NT, Medinas-Santos GM, Sarda-Rodriguez IL, Acosta-Martinez MA, Reyes-Matienzo R, Coviella-Artime JM, Morffi-Cinta I, Martínez-Pérez M, Valera-Fernández R, Garcés-Hechavarría A, Martínez-Bedoya D, Garrido-Arteaga R, Cardoso-SanJorge F, Ramírez-Gonzalez U, Quintero-Moreno L, Ontivero-Pino I, Martínez-Rivera R, Guillén-Obregón B, Lora-García J, Medina-Nápoles M, Espi-Ávila J, Fontanies-Fernández M, Domínguez-Pentón YR, Bergado-Baez G, Pi-Estopiñán F, Ojito-Magaz E, Rodríguez M, Cruz-Sui O, García-Montero M, Dubed-Echevarría M, García-López E, Galano-Frutos E, Perez-Perez A, Morales-Ruano S, Brito-Pascual I, Amoroto M, Arteaga-García A. 2022. Safety and immunogenicity of anti-SARS-CoV-2 heterologous scheme with SOBERANA 02 and SOBERANA Plus vaccines: Phase IIb clinical trial in adults. Med 3:760–773.e5.
1324.
Limonta-Fernández M, Chinea-Santiago G, Martín-Dunn AM, Gonzalez-Roche D, Bequet-Romero M, Marquez-Perera G, González-Moya I, Canaan-Haden-Ayala C, Cabrales-Rico A, Espinosa-Rodríguez LA, Ramos-Gómez Y, Andujar-Martínez I, González-López LJ, de la Iglesia MP, Zamora-Sanchez J, Cruz-Sui O, Lemos-Pérez G, Cabrera-Herrera G, Valdes-Hernández J, Martinez-Diaz E, Pimentel-Vazquez E, Ayala-Avila M, Guillén-Nieto G. 2022. An engineered SARS-CoV-2 receptor-binding domain produced in Pichia pastoris as a candidate vaccine antigen. New Biotechnology 72:11–21.
1325.
Hernández-Bernal F, Ricardo-Cobas MC, Martín-Bauta Y, Navarro-Rodríguez Z, Piñera-Martínez M, Quintana-Guerra J, Urrutia-Pérez K, Urrutia-Pérez K, Chávez-Chong CO, Azor-Hernández JL, Rodríguez-Reinoso JL, Lobaina-Lambert L, Colina-Ávila E, Bizet-Almeida J, Rodríguez-Nuviola J, del Valle-Piñera S, Ramírez-Domínguez M, Tablada-Ferreiro E, Alonso-Valdés M, Lemos-Pérez G, Guillén-Nieto GE, Palenzuela-Díaz A, Noa-Romero E, Limonta-Fernández M, Fernández-Ávila JM, Ali-Mros NA, del Toro-Lahera L, Remedios-Reyes R, Ayala-Ávila M, Muzio-González VL. 2022. Safety, tolerability, and immunogenicity of a SARS-CoV-2 recombinant spike RBD protein vaccine: A randomised, double-blind, placebo-controlled, phase 1-2 clinical trial (ABDALA Study). eClinicalMedicine 46:101383.
Hernández-Bernal F, Ricardo-Cobas MC, Martín-Bauta Y, Rodríguez-Martínez E, Urrutia-Pérez K, Urrutia-Pérez K, Quintana-Guerra J, Navarro-Rodríguez Z, Piñera-Martínez M, Rodríguez-Reinoso JL, Chávez-Chong CO, Baladrón-Castrillo I, Melo-Suárez G, Batista-Izquierdo A, Pupo-Micó A, Mora-Betancourt R, Soler-Cano D, Bizet-Almeida J, Martínez-Rodríguez MC, Lobaina-Lambert L, Velázquez-Pérez VM, Soler-Díaz J, Blanco-Garrido Y, Laurencio-Vallina S, Meriño-Hechavarría T, Carmenaty-Campos N, Rodríguez-Montero E, Limonta-Fernández M, Alonso-Valdés M, Hernández-Rodríguez R, Pimentel-Vázquez E, Catasús-Álvarez KM, Cabrera-Núñez MV, Ayala-Ávila M, Muzio-González VL. 2022. A phase 3, randomised, double-blind, placebo-controlled clinical trial for adult evaluation of the efficacy and safety of a SARS-CoV-2 recombinant spike RBD protein vaccine (ABDALA-3 Study). Cold Spring Harbor Laboratory.
Harapan H, Anwar S, Yufika A, Sharun K, Gachabayov M, Fahriani M, Husnah M, Raad R, Abdalla RY, Adam RY, Khiri NME, Ismaeil MIH, Ismail AY, Kacem W, Dahman NBH, Teyeb Z, Aloui K, Hafsi M, Ferjani M, Deeb DA, Emad D, Abbas KS, Monib FA, Sami FS, Subramaniam R, Panchawagh S, Anandu S, Haque MA, Ferreto LED, Briones MFC, Morales RBI, Díaz SAL, Aburto JTO, Rojas JET, Balogun EO, Enitan SS, Yomi AR, Durosinmi A, Ezigbo ED, Adejumo EN, Babadi E, Kakemam E, Malik NI, Ullah I, Rosiello DF, Emran TB, Wendt GW, Arab-Zozani M, Wagner AL, Mudatsir M. 2021. Vaccine hesitancy among communities in ten countries in Asia, Africa, and South America during the COVID-19 pandemic. Pathogens and Global Health 116:236–243.
Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, Joosten LAB, van der Meer JWM, Mhlanga MM, Mulder WJM, Riksen NP, Schlitzer A, Schultze JL, Stabell Benn C, Sun JC, Xavier RJ, Latz E. 2020. Defining trained immunity and its role in health and disease. Nature Reviews Immunology 20:375–388.
Reducing Health Care Workers Absenteeism in Covid-19 Pandemic Through BCG Vaccine - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04328441. Retrieved 8 February 2021.
COVID-19: BCG As Therapeutic Vaccine, Transmission Limitation, and Immunoglobulin Enhancement - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04369794. Retrieved 8 February 2021.
1446.
Using BCG Vaccine to Protect Health Care Workers in the COVID-19 Pandemic - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04373291. Retrieved 8 February 2021.
Study to Assess VPM1002 in Reducing Healthcare Professionals' Absenteeism in COVID-19 Pandemic - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04387409. Retrieved 8 February 2021.
Study to Assess VPM1002 in Reducing Hospital Admissions and/or Severe Respiratory Infectious Diseases in Elderly in COVID-19 Pandemic - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04435379. Retrieved 8 February 2021.
1453.
Efficacy and Safety of VPM1002 in Reducing SARS-CoV-2 (COVID-19) Infection Rate and Severity - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04439045. Retrieved 8 February 2021.
1454.
Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, Damoraki G, Gkavogianni T, Adami M-E, Katsaounou P, Ntaganou M, Kyriakopoulou M, Dimopoulos G, Koutsodimitropoulos I, Velissaris D, Koufargyris P, Karageorgos A, Katrini K, Lekakis V, Lupse M, Kotsaki A, Renieris G, Theodoulou D, Panou V, Koukaki E, Koulouris N, Gogos C, Koutsoukou A. 2020. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host & Microbe 27:992–1000.e3.
Voysey M, Clemens SAC, Madhi SA, Weckx LY, Folegatti PM, Aley PK, Angus B, Baillie VL, Barnabas SL, Bhorat QE, Bibi S, Briner C, Cicconi P, Collins AM, Colin-Jones R, Cutland CL, Darton TC, Dheda K, Duncan CJA, Emary KRW, Ewer KJ, Fairlie L, Faust SN, Feng S, Ferreira DM, Finn A, Goodman AL, Green CM, Green CA, Heath PT, Hill C, Hill H, Hirsch I, Hodgson SHC, Izu A, Jackson S, Jenkin D, Joe CCD, Kerridge S, Koen A, Kwatra G, Lazarus R, Lawrie AM, Lelliott A, Libri V, Lillie PJ, Mallory R, Mendes AVA, Milan EP, Minassian AM, McGregor A, Morrison H, Mujadidi YF, Nana A, O’Reilly PJ, Padayachee SD, Pittella A, Plested E, Pollock KM, Ramasamy MN, Rhead S, Schwarzbold AV, Singh N, Smith A, Song R, Snape MD, Sprinz E, Sutherland RK, Tarrant R, Thomson EC, Török ME, Toshner M, Turner DPJ, Vekemans J, Villafana TL, Watson MEE, Williams CJ, Douglas AD, Hill AVS, Lambe T, Gilbert SC, Pollard AJ, Aban M, Abayomi F, Abeyskera K, Aboagye J, Adam M, Adams K, Adamson J, Adelaja YA, Adewetan G, Adlou S, Ahmed K, Akhalwaya Y, Akhalwaya S, Alcock A, Ali A, Allen ER, Allen L, Almeida TCDSC, Alves MPS, Amorim F, Andritsou F, Anslow R, Appleby M, Arbe-Barnes EH, Ariaans MP, Arns B, Arruda L, Azi P, Azi L, Babbage G, Bailey C, Baker KF, Baker M, Baker N, Baker P, Baldwin L, Baleanu I, Bandeira D, Bara A, Barbosa MAS, Barker D, Barlow GD, Barnes E, Barr AS, Barrett JR, Barrett J, Bates L, Batten A, Beadon K, Beales E, Beckley R, Belij-Rammerstorfer S, Bell J, Bellamy D, Bellei N, Belton S, Berg A, Bermejo L, Berrie E, Berry L, Berzenyi D, Beveridge A, Bewley KR, Bexhell H, Bhikha S, Bhorat AE, Bhorat ZE, Bijker E, Birch G, Birch S, Bird A, Bird O, Bisnauthsing K, Bittaye M, Blackstone K, Blackwell L, Bletchly H, Blundell CL, Blundell SR, Bodalia P, Boettger BC, Bolam E, Boland E, Bormans D, Borthwick N, Bowring F, Boyd A, Bradley P, Brenner T, Brown P, Brown C, Brown-O'Sullivan C, Bruce S, Brunt E, Buchan R, Budd W, Bulbulia YA, Bull M, Burbage J, Burhan H, Burn A, Buttigieg KR, Byard N, Cabera Puig I, Calderon G, Calvert A, Camara S, Cao M, Cappuccini F, Cardoso JR, Carr M, Carroll MW, Carson-Stevens A, Carvalho Y de M, Carvalho JAM, Casey HR, Cashen P, Castro T, Castro LC, Cathie K, Cavey A, Cerbino-Neto J, Chadwick J, Chapman D, Charlton S, Chelysheva I, Chester O, Chita S, Cho J-S, Cifuentes L, Clark E, Clark M, Clarke A, Clutterbuck EA, Collins SLK, Conlon CP, Connarty S, Coombes N, Cooper C, Cooper R, Cornelissen L, Corrah T, Cosgrove C, Cox T, Crocker WEM, Crosbie S, Cullen L, Cullen D, Cunha DRMF, Cunningham C, Cuthbertson FC, Da Guarda SNF, da Silva LP, Damratoski BE, Danos Z, Dantas MTDC, Darroch P, Datoo MS, Datta C, Davids M, Davies SL, Davies H, Davis E, Davis J, Davis J, De Nobrega MMD, De Oliveira Kalid LM, Dearlove D, Demissie T, Desai A, Di Marco S, Di Maso C, Dinelli MIS, Dinesh T, Docksey C, Dold C, Dong T, Donnellan FR, Dos Santos T, dos Santos TG, Dos Santos EP, Douglas N, Downing C, Drake J, Drake-Brockman R, Driver K, Drury R, Dunachie SJ, Durham BS, Dutra L, Easom NJW, van Eck S, Edwards M, Edwards NJ, El Muhanna OM, Elias SC, Elmore M, English M, Esmail A, Essack YM, Farmer E, Farooq M, Farrar M, Farrugia L, Faulkner B, Fedosyuk S, Felle S, Feng S, Ferreira Da Silva C, Field S, Fisher R, Flaxman A, Fletcher J, Fofie H, Fok H, Ford KJ, Fowler J, Fraiman PHA, Francis E, Franco MM, Frater J, Freire MSM, Fry SH, Fudge S, Furze J, Fuskova M, Galian-Rubio P, Galiza E, Garlant H, Gavrila M, Geddes A, Gibbons KA, Gilbride C, Gill H, Glynn S, Godwin K, Gokani K, Goldoni UC, Goncalves M, Gonzalez IGS, Goodwin J, Goondiwala A, Gordon-Quayle K, Gorini G, Grab J, Gracie L, Greenland M, Greenwood N, Greffrath J, Groenewald MM, Grossi L, Gupta G, Hackett M, Hallis B, Hamaluba M, Hamilton E, Hamlyn J, Hammersley D, Hanrath AT, Hanumunthadu B, Harris SA, Harris C, Harris T, Harrison TD, Harrison D, Hart TC, Hartnell B, Hassan S, Haughney J, Hawkins S, Hay J, Head I, Henry J, Hermosin Herrera M, Hettle DB, Hill J, Hodges G, Horne E, Hou MM, Houlihan C, Howe E, Howell N, Humphreys J, Humphries HE, Hurley K, Huson C, Hyder-Wright A, Hyams C, Ikram S, Ishwarbhai A, Ivan M, Iveson P, Iyer V, Jackson F, De Jager J, Jaumdally S, Jeffers H, Jesudason N, Jones B, Jones K, Jones E, Jones C, Jorge MR, Jose A, Joshi A, Júnior EAMS, Kadziola J, Kailath R, Kana F, Karampatsas K, Kasanyinga M, Keen J, Kelly EJ, Kelly DM, Kelly D, Kelly S, Kerr D, Kfouri R de Á, Khan L, Khozoee B, Kidd S, Killen A, Kinch J, Kinch P, King LDW, King TB, Kingham L, Klenerman P, Knapper F, Knight JC, Knott D, Koleva S, Lang M, Lang G, Larkworthy CW, Larwood JPJ, Law R, Lazarus EM, Leach A, Lees EA, Lemm N-M, Lessa A, Leung S, Li Y, Lias AM, Liatsikos K, Linder A, Lipworth S, Liu S, Liu X, Lloyd A, Lloyd S, Loew L, Lopez Ramon R, Lora L, Lowthorpe V, Luz K, MacDonald JC, MacGregor G, Madhavan M, Mainwaring DO, Makambwa E, Makinson R, Malahleha M, Malamatsho R, Mallett G, Mansatta K, Maoko T, Mapetla K, Marchevsky NG, Marinou S, Marlow E, Marques GN, Marriott P, Marshall RP, Marshall JL, Martins FJ, Masenya M, Masilela M, Masters SK, Mathew M, Matlebjane H, Matshidiso K, Mazur O, Mazzella A, McCaughan H, McEwan J, McGlashan J, McInroy L, McIntyre Z, McLenaghan D, McRobert N, McSwiggan S, Megson C, Mehdipour S, Meijs W, Mendonça RNÁ, Mentzer AJ, Mirtorabi N, Mitton C, Mnyakeni S, Moghaddas F, Molapo K, Moloi M, Moore M, Moraes-Pinto MI, Moran M, Morey E, Morgans R, Morris S, Morris S, Morris HC, Morselli F, Morshead G, Morter R, Mottal L, Moultrie A, Moya N, Mpelembue M, Msomi S, Mugodi Y, Mukhopadhyay E, Muller J, Munro A, Munro C, Murphy S, Mweu P, Myasaki CH, Naik G, Naker K, Nastouli E, Nazir A, Ndlovu B, Neffa F, Njenga C, Noal H, Noé A, Novaes G, Nugent FL, Nunes G, O'Brien K, O'Connor D, Odam M, Oelofse S, Oguti B, Olchawski V, Oldfield NJ, Oliveira MG, Oliveira C, Oosthuizen A, O'Reilly P, Osborne P, Owen DRJ, Owen L, Owens D, Owino N, Pacurar M, Paiva BVB, Palhares EMF, Palmer S, Parkinson S, Parracho HMRT, Parsons K, Patel D, Patel B, Patel F, Patel K, Patrick-Smith M, Payne RO, Peng Y, Penn EJ, Pennington A, Peralta Alvarez MP, Perring J, Perry N, Perumal R, Petkar S, Philip T, Phillips DJ, Phillips J, Phohu MK, Pickup L, Pieterse S, Piper J, Pipini D, Plank M, Du Plessis J, Pollard S, Pooley J, Pooran A, Poulton I, Powers C, Presa FB, Price DA, Price V, Primeira M, Proud PC, Provstgaard-Morys S, Pueschel S, Pulido D, Quaid S, Rabara R, Radford A, Radia K, Rajapaska D, Rajeswaran T, Ramos ASF, Ramos Lopez F, Rampling T, Rand J, Ratcliffe H, Rawlinson T, Rea D, Rees B, Reiné J, Resuello-Dauti M, Reyes Pabon E, Ribiero CM, Ricamara M, Richter A, Ritchie N, Ritchie AJ, Robbins AJ, Roberts H, Robinson RE, Robinson H, Rocchetti TT, Rocha BP, Roche S, Rollier C, Rose L, Ross Russell AL, Rossouw L, Royal S, Rudiansyah I, Ruiz S, Saich S, Sala C, Sale J, Salman AM, Salvador N, Salvador S, Sampaio M, Samson AD, Sanchez-Gonzalez A, Sanders H, Sanders K, Santos E, Santos Guerra MFS, Satti I, Saunders JE, Saunders C, Sayed A, Schim van der Loeff I, Schmid AB, Schofield E, Screaton G, Seddiqi S, Segireddy RR, Senger R, Serrano S, Shah R, Shaik I, Sharpe HE, Sharrocks K, Shaw R, Shea A, Shepherd A, Shepherd JG, Shiham F, Sidhom E, Silk SE, da Silva Moraes AC, Silva-Junior G, Silva-Reyes L, Silveira AD, Silveira MBV, Sinha J, Skelly DT, Smith DC, Smith N, Smith HE, Smith DJ, Smith CC, Soares A, Soares T, Solórzano C, Sorio GL, Sorley K, Sosa-Rodriguez T, Souza CMCDL, Souza BSDF, Souza AR, Spencer AJ, Spina F, Spoors L, Stafford L, Stamford I, Starinskij I, Stein R, Steven J, Stockdale L, Stockwell LV, Strickland LH, Stuart AC, Sturdy A, Sutton N, Szigeti A, Tahiri-Alaoui A, Tanner R, Taoushanis C, Tarr AW, Taylor K, Taylor U, Taylor IJ, Taylor J, te Water Naude R, Themistocleous Y, Themistocleous A, Thomas M, Thomas K, Thomas TM, Thombrayil A, Thompson F, Thompson A, Thompson K, Thompson A, Thomson J, Thornton-Jones V, Tighe PJ, Tinoco LA, Tiongson G, Tladinyane B, Tomasicchio M, Tomic A, Tonks S, Towner J, Tran N, Tree J, Trillana G, Trinham C, Trivett R, Truby A, Tsheko BL, Turabi A, Turner R, Turner C, Ulaszewska M, Underwood BR, Varughese R, Verbart D, Verheul M, Vichos I, Vieira T, Waddington CS, Walker L, Wallis E, Wand M, Warbick D, Wardell T, Warimwe G, Warren SC, Watkins B, Watson E, Webb S, Webb-Bridges A, Webster A, Welch J, Wells J, West A, White C, White R, Williams P, Williams RL, Winslow R, Woodyer M, Worth AT, Wright D, Wroblewska M, Yao A, Zimmer R, Zizi D, Zuidewind P. 2021. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. The Lancet 397:99–111.
Safety, Tolerability and Immunogenicity of INO-4800 for COVID-19 in Healthy Volunteers - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04336410. Retrieved 8 February 2021.
Tebas P, Yang S, Boyer JD, Reuschel EL, Patel A, Christensen-Quick A, Andrade VM, Morrow MP, Kraynyak K, Agnes J, Purwar M, Sylvester A, Pawlicki J, Gillespie E, Maricic I, Zaidi FI, Kim KY, Dia Y, Frase D, Pezzoli P, Schultheis K, Smith TRF, Ramos SJ, McMullan T, Buttigieg K, Carroll MW, Ervin J, Diehl MC, Blackwood E, Mammen MP, Lee J, Dallas MJ, Brown AS, Shea JE, Kim JJ, Weiner DB, Broderick KE, Humeau LM. 2021. Safety and immunogenicity of INO-4800 DNA vaccine against SARS-CoV-2: A preliminary report of an open-label, Phase 1 clinical trial. EClinicalMedicine 31:100689.
Kraynyak KA, Blackwood E, Agnes J, Tebas P, Giffear M, Amante D, Reuschel EL, Purwar M, Christensen-Quick A, Liu N, Andrade VM, Diehl MC, Wani S, Lupicka M, Sylvester A, Morrow MP, Pezzoli P, McMullan T, Kulkarni AJ, Zaidi FI, Frase D, Liaw K, Smith TRF, Ramos SJ, Ervin J, Adams M, Lee J, Dallas M, Shah Brown A, Shea JE, Kim JJ, Weiner DB, Broderick KE, Humeau LM, Boyer JD, Mammen MP Jr. 2022. SARS-CoV-2 DNA Vaccine INO-4800 Induces Durable Immune Responses Capable of Being Boosted in a Phase 1 Open-Label Trial. The Journal of Infectious Diseases 225:1923–1932.
Reed CC, Schultheis K, Andrade VM, Kalia R, Tur J, Schouest B, Elwood D, Walters JN, Maricic I, Doan A, Vazquez M, Eblimit Z, Pezzoli P, Amante D, Porto M, Narvaez B, Lok M, Spence B, Bradette H, Horn H, Yang M, Fader J, Ferrer R, Weiner DB, Kar S, Kim JJ, Humeau LM, Ramos SJ, Smith TRF, Broderick KE. 2021. Design, immunogenicity and efficacy of a Pan-SARS-CoV-2 synthetic DNA vaccine. Cold Spring Harbor Laboratory.
Bliss CM, Drammeh A, Bowyer G, Sanou GS, Jagne YJ, Ouedraogo O, Edwards NJ, Tarama C, Ouedraogo N, Ouedraogo M, Njie-Jobe J, Diarra A, Afolabi MO, Tiono AB, Yaro JB, Adetifa UJ, Hodgson SH, Anagnostou NA, Roberts R, Duncan CJA, Cortese R, Viebig NK, Leroy O, Lawrie AM, Flanagan KL, Kampmann B, Imoukhuede EB, Sirima SB, Bojang K, Hill AVS, Nébié I, Ewer KJ. 2017. Viral Vector Malaria Vaccines Induce High-Level T Cell and Antibody Responses in West African Children and Infants. Molecular Therapy 25:547–559.
1544.
Li S, Locke E, Bruder J, Clarke D, Doolan DL, Havenga MJE, Hill AVS, Liljestrom P, Monath TP, Naim HY, Ockenhouse C, Tang DC, Van Kampen KR, Viret J-F, Zavala F, Dubovsky F. 2007. Viral vectors for malaria vaccine development. Vaccine 25:2567–2574.
1545.
Ledgerwood JE, DeZure AD, Stanley DA, Coates EE, Novik L, Enama ME, Berkowitz NM, Hu Z, Joshi G, Ploquin A, Sitar S, Gordon IJ, Plummer SA, Holman LA, Hendel CS, Yamshchikov G, Roman F, Nicosia A, Colloca S, Cortese R, Bailer RT, Schwartz RM, Roederer M, Mascola JR, Koup RA, Sullivan NJ, Graham BS. 2017. Chimpanzee Adenovirus Vector Ebola Vaccine. N Engl J Med 376:928–938.
van Doremalen N, Lambe T, Spencer A, Belij-Rammerstorfer S, Purushotham JN, Port JR, Avanzato VA, Bushmaker T, Flaxman A, Ulaszewska M, Feldmann F, Allen ER, Sharpe H, Schulz J, Holbrook M, Okumura A, Meade-White K, Pérez-Pérez L, Edwards NJ, Wright D, Bissett C, Gilbride C, Williamson BN, Rosenke R, Long D, Ishwarbhai A, Kailath R, Rose L, Morris S, Powers C, Lovaglio J, Hanley PW, Scott D, Saturday G, de Wit E, Gilbert SC, Munster VJ. 2020. ChAdOx1 nCoV-19 vaccine prevents SARS-CoV-2 pneumonia in rhesus macaques. Nature 586:578–582.
1557.
Folegatti PM, Ewer KJ, Aley PK, Angus B, Becker S, Belij-Rammerstorfer S, Bellamy D, Bibi S, Bittaye M, Clutterbuck EA, Dold C, Faust SN, Finn A, Flaxman AL, Hallis B, Heath P, Jenkin D, Lazarus R, Makinson R, Minassian AM, Pollock KM, Ramasamy M, Robinson H, Snape M, Tarrant R, Voysey M, Green C, Douglas AD, Hill AVS, Lambe T, Gilbert SC, Pollard AJ, Aboagye J, Adams K, Ali A, Allen E, Allison JL, Anslow R, Arbe-Barnes EH, Babbage G, Baillie K, Baker M, Baker N, Baker P, Baleanu I, Ballaminut J, Barnes E, Barrett J, Bates L, Batten A, Beadon K, Beckley R, Berrie E, Berry L, Beveridge A, Bewley KR, Bijker EM, Bingham T, Blackwell L, Blundell CL, Bolam E, Boland E, Borthwick N, Bower T, Boyd A, Brenner T, Bright PD, Brown-O'Sullivan C, Brunt E, Burbage J, Burge S, Buttigieg KR, Byard N, Cabera Puig I, Calvert A, Camara S, Cao M, Cappuccini F, Carr M, Carroll MW, Carter V, Cathie K, Challis RJ, Charlton S, Chelysheva I, Cho J-S, Cicconi P, Cifuentes L, Clark H, Clark E, Cole T, Colin-Jones R, Conlon CP, Cook A, Coombes NS, Cooper R, Cosgrove CA, Coy K, Crocker WEM, Cunningham CJ, Damratoski BE, Dando L, Datoo MS, Davies H, De Graaf H, Demissie T, Di Maso C, Dietrich I, Dong T, Donnellan FR, Douglas N, Downing C, Drake J, Drake-Brockman R, Drury RE, Dunachie SJ, Edwards NJ, Edwards FDL, Edwards CJ, Elias SC, Elmore MJ, Emary KRW, English MR, Fagerbrink S, Felle S, Feng S, Field S, Fixmer C, Fletcher C, Ford KJ, Fowler J, Fox P, Francis E, Frater J, Furze J, Fuskova M, Galiza E, Gbesemete D, Gilbride C, Godwin K, Gorini G, Goulston L, Grabau C, Gracie L, Gray Z, Guthrie LB, Hackett M, Halwe S, Hamilton E, Hamlyn J, Hanumunthadu B, Harding I, Harris SA, Harris A, Harrison D, Harrison C, Hart TC, Haskell L, Hawkins S, Head I, Henry JA, Hill J, Hodgson SHC, Hou MM, Howe E, Howell N, Hutlin C, Ikram S, Isitt C, Iveson P, Jackson S, Jackson F, James SW, Jenkins M, Jones E, Jones K, Jones CE, Jones B, Kailath R, Karampatsas K, Keen J, Kelly S, Kelly D, Kerr D, Kerridge S, Khan L, Khan U, Killen A, Kinch J, King TB, King L, King J, Kingham-Page L, Klenerman P, Knapper F, Knight JC, Knott D, Koleva S, Kupke A, Larkworthy CW, Larwood JPJ, Laskey A, Lawrie AM, Lee A, Ngan Lee KY, Lees EA, Legge H, Lelliott A, Lemm N-M, Lias AM, Linder A, Lipworth S, Liu X, Liu S, Lopez Ramon R, Lwin M, Mabesa F, Madhavan M, Mallett G, Mansatta K, Marcal I, Marinou S, Marlow E, Marshall JL, Martin J, McEwan J, McInroy L, Meddaugh G, Mentzer AJ, Mirtorabi N, Moore M, Moran E, Morey E, Morgan V, Morris SJ, Morrison H, Morshead G, Morter R, Mujadidi YF, Muller J, Munera-Huertas T, Munro C, Munro A, Murphy S, Munster VJ, Mweu P, Noé A, Nugent FL, Nuthall E, O'Brien K, O'Connor D, Oguti B, Oliver JL, Oliveira C, O'Reilly PJ, Osborn M, Osborne P, Owen C, Owens D, Owino N, Pacurar M, Parker K, Parracho H, Patrick-Smith M, Payne V, Pearce J, Peng Y, Peralta Alvarez MP, Perring J, Pfafferott K, Pipini D, Plested E, Pluess-Hall H, Pollock K, Poulton I, Presland L, Provstgaard-Morys S, Pulido D, Radia K, Ramos Lopez F, Rand J, Ratcliffe H, Rawlinson T, Rhead S, Riddell A, Ritchie AJ, Roberts H, Robson J, Roche S, Rohde C, Rollier CS, Romani R, Rudiansyah I, Saich S, Sajjad S, Salvador S, Sanchez Riera L, Sanders H, Sanders K, Sapaun S, Sayce C, Schofield E, Screaton G, Selby B, Semple C, Sharpe HR, Shaik I, Shea A, Shelton H, Silk S, Silva-Reyes L, Skelly DT, Smee H, Smith CC, Smith DJ, Song R, Spencer AJ, Stafford E, Steele A, Stefanova E, Stockdale L, Szigeti A, Tahiri-Alaoui A, Tait M, Talbot H, Tanner R, Taylor IJ, Taylor V, Te Water Naude R, Thakur N, Themistocleous Y, Themistocleous A, Thomas M, Thomas TM, Thompson A, Thomson-Hill S, Tomlins J, Tonks S, Towner J, Tran N, Tree JA, Truby A, Turkentine K, Turner C, Turner N, Turner S, Tuthill T, Ulaszewska M, Varughese R, Van Doremalen N, Veighey K, Verheul MK, Vichos I, Vitale E, Walker L, Watson MEE, Welham B, Wheat J, White C, White R, Worth AT, Wright D, Wright S, Yao XL, Yau Y. 2020. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. The Lancet 396:467–478.
Barouch DH, Kik SV, Weverling GJ, Dilan R, King SL, Maxfield LF, Clark S, Ng’ang’a D, Brandariz KL, Abbink P, Sinangil F, de Bruyn G, Gray GE, Roux S, Bekker L-G, Dilraj A, Kibuuka H, Robb ML, Michael NL, Anzala O, Amornkul PN, Gilmour J, Hural J, Buchbinder SP, Seaman MS, Dolin R, Baden LR, Carville A, Mansfield KG, Pau MG, Goudsmit J. 2011. International seroepidemiology of adenovirus serotypes 5, 26, 35, and 48 in pediatric and adult populations. Vaccine 29:5203–5209.
1560.
Office USGA. Operation Warp Speed: Accelerated COVID-19 Vaccine Development Status and Efforts to Address Manufacturing Challenges. https://www.gao.gov/products/gao-21-319. Retrieved 5 December 2022.
Sadoff J, Le Gars M, Shukarev G, Heerwegh D, Truyers C, de Groot AM, Stoop J, Tete S, Van Damme W, Leroux-Roels I, Berghmans P-J, Kimmel M, Van Damme P, de Hoon J, Smith W, Stephenson KE, De Rosa SC, Cohen KW, McElrath MJ, Cormier E, Scheper G, Barouch DH, Hendriks J, Struyf F, Douoguih M, Van Hoof J, Schuitemaker H. 2021. Interim Results of a Phase 1–2a Trial of Ad26.COV2.S Covid-19 Vaccine. N Engl J Med 384:1824–1835.
1563.
Mercado NB, Zahn R, Wegmann F, Loos C, Chandrashekar A, Yu J, Liu J, Peter L, McMahan K, Tostanoski LH, He X, Martinez DR, Rutten L, Bos R, van Manen D, Vellinga J, Custers J, Langedijk JP, Kwaks T, Bakkers MJG, Zuijdgeest D, Rosendahl Huber SK, Atyeo C, Fischinger S, Burke JS, Feldman J, Hauser BM, Caradonna TM, Bondzie EA, Dagotto G, Gebre MS, Hoffman E, Jacob-Dolan C, Kirilova M, Li Z, Lin Z, Mahrokhian SH, Maxfield LF, Nampanya F, Nityanandam R, Nkolola JP, Patel S, Ventura JD, Verrington K, Wan H, Pessaint L, Van Ry A, Blade K, Strasbaugh A, Cabus M, Brown R, Cook A, Zouantchangadou S, Teow E, Andersen H, Lewis MG, Cai Y, Chen B, Schmidt AG, Reeves RK, Baric RS, Lauffenburger DA, Alter G, Stoffels P, Mammen M, Van Hoof J, Schuitemaker H, Barouch DH. 2020. Single-shot Ad26 vaccine protects against SARS-CoV-2 in rhesus macaques. Nature 586:583–588.
1564.
Tostanoski LH, Wegmann F, Martinot AJ, Loos C, McMahan K, Mercado NB, Yu J, Chan CN, Bondoc S, Starke CE, Nekorchuk M, Busman-Sahay K, Piedra-Mora C, Wrijil LM, Ducat S, Custers J, Atyeo C, Fischinger S, Burke JS, Feldman J, Hauser BM, Caradonna TM, Bondzie EA, Dagotto G, Gebre MS, Jacob-Dolan C, Lin Z, Mahrokhian SH, Nampanya F, Nityanandam R, Pessaint L, Porto M, Ali V, Benetiene D, Tevi K, Andersen H, Lewis MG, Schmidt AG, Lauffenburger DA, Alter G, Estes JD, Schuitemaker H, Zahn R, Barouch DH. 2020. Ad26 vaccine protects against SARS-CoV-2 severe clinical disease in hamsters. Nat Med 26:1694–1700.
1565.
Solforosi L, Kuipers H, Huber SKR, van der Lubbe JEM, Dekking L, Czapska-Casey DN, Gil AI, Baert MRM, Drijver J, Vaneman J, van Huizen E, Choi Y, Vreugdenhil J, Kroos S, de Wilde AH, Kourkouta E, Custers J, Dalebout TJ, Myeni SK, Kikkert M, Snijder EJ, Barouch DH, Böszörményi KP, Stammes MA, Kondova I, Verschoor EJ, Verstrepen BE, Koopman G, Mooij P, Bogers WMJM, van Heerden M, Muchene L, Tolboom JTBM, Roozendaal R, Schuitemaker H, Wegmann F, Zahn RC. 2020. Immunogenicity and protective efficacy of one- and two-dose regimens of the Ad26.COV2.S COVID-19 vaccine candidate in adult and aged rhesus macaques. Cold Spring Harbor Laboratory.
Cohen J. 2020. Russia’s claim of a successful COVID-19 vaccine doesn’t pass the ‘smell test,’ critics say. Science https://doi.org/10.1126/science.abf6791.
1579.
Callaway E. 2020. Russia announces positive COVID-vaccine results from controversial trial. Nature https://doi.org/10.1038/d41586-020-03209-0.
Phillips N, Cyranoski D, Mallapaty S. 2020. A leading coronavirus vaccine trial is on hold: scientists react. Nature https://doi.org/10.1038/d41586-020-02594-w.
Baker AT, Boyd RJ, Sarkar D, Teijeira-Crespo A, Chan CK, Bates E, Waraich K, Vant J, Wilson E, Truong CD, Lipka-Lloyd M, Fromme P, Vermaas J, Williams D, Machiesky L, Heurich M, Nagalo BM, Coughlan L, Umlauf S, Chiu P-L, Rizkallah PJ, Cohen TS, Parker AL, Singharoy A, Borad MJ. 2021. ChAdOx1 interacts with CAR and PF4 with implications for thrombosis with thrombocytopenia syndrome. Sci Adv 7.
Study in Healthy Adults to Evaluate Gene Activation After Vaccination With GlaxoSmithKline (GSK) Biologicals' Candidate Tuberculosis (TB) Vaccine GSK 692342 - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01669096. Retrieved 8 February 2021.
Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M, Levantovsky R, Malle L, Moreira A, Park MD, Pia L, Risson E, Saffern M, Salomé B, Esai Selvan M, Spindler MP, Tan J, van der Heide V, Gregory JK, Alexandropoulos K, Bhardwaj N, Brown BD, Greenbaum B, Gümüş ZH, Homann D, Horowitz A, Kamphorst AO, Curotto de Lafaille MA, Mehandru S, Merad M, Samstein RM, Agrawal M, Aleynick M, Belabed M, Brown M, Casanova-Acebes M, Catalan J, Centa M, Charap A, Chan A, Chen ST, Chung J, Bozkus CC, Cody E, Cossarini F, Dalla E, Fernandez N, Grout J, Ruan DF, Hamon P, Humblin E, Jha D, Kodysh J, Leader A, Lin M, Lindblad K, Lozano-Ojalvo D, Lubitz G, Magen A, Mahmood Z, Martinez-Delgado G, Mateus-Tique J, Meritt E, Moon C, Noel J, O’Donnell T, Ota M, Plitt T, Pothula V, Redes J, Reyes Torres I, Roberto M, Sanchez-Paulete AR, Shang J, Schanoski AS, Suprun M, Tran M, Vaninov N, Wilk CM, Aguirre-Ghiso J, Bogunovic D, Cho J, Faith J, Grasset E, Heeger P, Kenigsberg E, Krammer F, Laserson U. 2020. Immunology of COVID-19: Current State of the Science. Immunity 52:910–941.
Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB, McGettigan J, Khetan S, Segall N, Solis J, Brosz A, Fierro C, Schwartz H, Neuzil K, Corey L, Gilbert P, Janes H, Follmann D, Marovich M, Mascola J, Polakowski L, Ledgerwood J, Graham BS, Bennett H, Pajon R, Knightly C, Leav B, Deng W, Zhou H, Han S, Ivarsson M, Miller J, Zaks T. 2020. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. New England Journal of Medicine NEJMoa2035389.
Andrews N, Stowe J, Kirsebom F, Toffa S, Rickeard T, Gallagher E, Gower C, Kall M, Groves N, O’Connell A-M, Simons D, Blomquist PB, Zaidi A, Nash S, Iwani Binti Abdul Aziz N, Thelwall S, Dabrera G, Myers R, Amirthalingam G, Gharbia S, Barrett JC, Elson R, Ladhani SN, Ferguson N, Zambon M, Campbell CNJ, Brown K, Hopkins S, Chand M, Ramsay M, Lopez Bernal J. 2022. Covid-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. N Engl J Med 386:1532–1546.
Abu Mouch S, Roguin A, Hellou E, Ishai A, Shoshan U, Mahamid L, Zoabi M, Aisman M, Goldschmid N, Berar Yanay N. 2021. Myocarditis following COVID-19 mRNA vaccination. Vaccine 39:3790–3793.
Mevorach D, Anis E, Cedar N, Bromberg M, Haas EJ, Nadir E, Olsha-Castell S, Arad D, Hasin T, Levi N, Asleh R, Amir O, Meir K, Cohen D, Dichtiar R, Novick D, Hershkovitz Y, Dagan R, Leitersdorf I, Ben-Ami R, Miskin I, Saliba W, Muhsen K, Levi Y, Green MS, Keinan-Boker L, Alroy-Preis S. 2021. Myocarditis after BNT162b2 mRNA Vaccine against Covid-19 in Israel. N Engl J Med 385:2140–2149.
1639.
Goddard K, Hanson KE, Lewis N, Weintraub E, Fireman B, Klein NP. 2022. Incidence of Myocarditis/Pericarditis Following mRNA COVID-19 Vaccination Among Children and Younger Adults in the United States. Ann Intern Med https://doi.org/10.7326/m22-2274.
1640.
Matta A, Kunadharaju R, Osman M, Jesme C, McMiller Z, Johnson EM, Matta D, Kallamadi R, Bande D. 2021. Clinical Presentation and Outcomes of Myocarditis Post mRNA Vaccination: A Meta-Analysis and Systematic Review. Cureus https://doi.org/10.7759/cureus.19240.
Hardt K, Vandebosch A, Sadoff J, Gars ML, Truyers C, Lowson D, Van Dromme I, Vingerhoets J, Kamphuis T, Scheper G, Ruiz-Guiñazú J, Faust SN, Spinner CD, Schuitemaker H, Van Hoof J, Douoguih M, Struyf F. 2022. Efficacy and Safety of a Booster Regimen of Ad26.COV2.S Vaccine against Covid-19. Cold Spring Harbor Laboratory.
Flaxman A, Marchevsky NG, Jenkin D, Aboagye J, Aley PK, Angus B, Belij-Rammerstorfer S, Bibi S, Bittaye M, Cappuccini F, Cicconi P, Clutterbuck EA, Davies S, Dejnirattisai W, Dold C, Ewer KJ, Folegatti PM, Fowler J, Hill AVS, Kerridge S, Minassian AM, Mongkolsapaya J, Mujadidi YF, Plested E, Ramasamy MN, Robinson H, Sanders H, Sheehan E, Smith H, Snape MD, Song R, Woods D, Screaton G, Gilbert SC, Voysey M, Pollard AJ, Lambe T, Adlou S, Aley R, Ali A, Anslow R, Baker M, Baker P, Barrett JR, Bates L, Beadon K, Beckley R, Bell J, Bellamy D, Beveridge A, Bissett C, Blackwell L, Bletchly H, Boyd A, Bridges-Webb A, Brown C, Byard N, Camara S, Cifuentes Gutierrez L, Collins AM, Cooper R, Crocker WEM, Darton TC, Davies H, Davies J, Demissie T, Di Maso C, Dinesh T, Donnellan FR, Douglas AD, Drake-Brockman R, Duncan CJA, Elias SC, Emary KRW, Ghulam Farooq M, Faust SN, Felle S, Ferreira D, Ferreira Da Silva C, Finn A, Ford KJ, Francis E, Furze J, Fuskova M, Galiza E, Gibertoni Cruz A, Godfrey L, Goodman AL, Green C, Green CA, Greenwood N, Harrison D, Hart TC, Hawkins S, Heath PT, Hill H, Hillson K, Horsington B, Hou MM, Howe E, Howell N, Joe C, Jones E, Kasanyinga M, Keen J, Kelly S, Kerr D, Khan L, Khozoee B, Kinch J, Kinch P, Koleva S, Kwok J, Larkworthy CW, Lawrie AM, Lazarus R, Lees EA, Li G, Libri V, Lillie PJ, Linder A, Long F, Lopez Ramon R, Mabbett R, Makinson R, Marinou S, Marlow E, Marshall JL, Mazur O, McEwan J, McGregor AC, Mokaya J, Morey E, Morshead G, Morter R, Muller J, Mweu P, Noristani R, Owino N, Polo Peralta Alvarez M, Platt A, Pollock KM, Poulton I, Provstgaard-Morys S, Pulido-Gomez D, Rajan M, Ramos Lopez F, Ritchie A, Roberts H, Rollier C, Rudiansyah I, Sanders K, Saunders JE, Seddiqi S, Sharpe HR, Shaw R, Silva-Reyes L, Singh N, Smith DJ, Smith CC, Smith A, Spencer AJ, Stuart ASV, Sutherland R, Szigeti A, Tang K, Thomas M, Thomas TM, Thompson A, Thomson EC, Török EM, Toshner M, Tran N, Trivett R, Turnbull I, Turner C, Turner DPJ, Ulaszewska M, Vichos I, Walker L, Watson ME, Whelan C, White R, Williams SJ, Williams CJA, Wright D, Yao A. 2021. Reactogenicity and immunogenicity after a late second dose or a third dose of ChAdOx1 nCoV-19 in the UK: a substudy of two randomised controlled trials (COV001 and COV002). The Lancet 398:981–990.
1649.
Regev-Yochay G, Gonen T, Gilboa M, Mandelboim M, Indenbaum V, Amit S, Meltzer L, Asraf K, Cohen C, Fluss R, Biber A, Nemet I, Kliker L, Joseph G, Doolman R, Mendelson E, Freedman LS, Harats D, Kreiss Y, Lustig Y. 2022. 4th Dose COVID mRNA Vaccines' Immunogenicity & Efficacy Against Omicron VOC. Cold Spring Harbor Laboratory.
Munro APS, Janani L, Cornelius V, Aley PK, Babbage G, Baxter D, Bula M, Cathie K, Chatterjee K, Dodd K, Enever Y, Gokani K, Goodman AL, Green CA, Harndahl L, Haughney J, Hicks A, van der Klaauw AA, Kwok J, Lambe T, Libri V, Llewelyn MJ, McGregor AC, Minassian AM, Moore P, Mughal M, Mujadidi YF, Murira J, Osanlou O, Osanlou R, Owens DR, Pacurar M, Palfreeman A, Pan D, Rampling T, Regan K, Saich S, Salkeld J, Saralaya D, Sharma S, Sheridan R, Sturdy A, Thomson EC, Todd S, Twelves C, Read RC, Charlton S, Hallis B, Ramsay M, Andrews N, Nguyen-Van-Tam JS, Snape MD, Liu X, Faust SN, Riordan A, Ustianowski A, Rogers CA, Hughes S, Longshaw L, Stockport J, Hughes R, Grundy L, Tudor Jones L, Guha A, Snashall E, Eadsforth T, Reeder S, Storton K, Munusamy M, Tandy B, Egbo A, Cox S, Ahmed NN, Shenoy A, Bousfield R, Wixted D, Gutteridge H, Mansfield B, Herbert C, Holliday K, Calderwood J, Barker D, Brandon J, Tulloch H, Colquhoun S, Thorp H, Radford H, Evans J, Baker H, Thorpe J, Batham S, Hailstone J, Phillips R, Kumar D, Westwell F, Makia F, Hopkins N, Barcella L, Mpelembue M, dabagh M, lang M, khan F, Adebambo O, Chita S, Corrah T, Whittington A, John L, Roche S, Wagstaff L, Farrier A, Bisnauthsing K, Serafimova T, Nanino E, Cooney E, Wilson-Goldsmith J, Nguyen H, Mazzella A, Jackson B, Aslam S, Bawa T, Broadhead S, Farooqi S, Piper J, Weighell R, Pickup L, Shamtally D, Domingo J, Kourampa E, Hale C, Gibney J, Stackpoole M, Rashid-Gardner Z, Lyon R, McDonnell C, Cole C, Stewart A, McMillan G, Savage M, Beckett H, Moorbey C, Desai A, Brown C, Naker K, Qureshi E, Trinham C, Sabine C, Moore S, Hurdover S, Justice E, Smith D, Plested E, Ferreira Da Silva C, White R, Robinson H, Cifuentes L, Morshead G, Drake-Brockman R, Kinch P, Kasanyinga M, Clutterbuck EA, Bibi S, Stuart AS, Shaw RH, Singh M, Champaneri T, Irwin M, Khan M, Kownacka A, Nabunjo M, Osuji C, Hladkiwskyj J, Galvin D, Patel G, Mouland J, Longhurst B, Moon M, Giddins B, Pereira Dias Alves C, Richmond L, Minnis C, Baryschpolec S, Elliott S, Fox L, Graham V, Baker N, Godwin K, Buttigieg K, Knight C, Brown P, Lall P, Shaik I, Chiplin E, Brunt E, Leung S, Allen L, Thomas S, Fraser S, Choi B, Gouriet J, Freedman A, Perkins J, Gowland A, Macdonald J, Seenan JP, Starinskij I, Seaton A, Peters E, Singh S, Gardside B, Bonnaud A, Davies C, Gordon E, Keenan S, Hall J, Wilkins S, Tasker S, James R, Seath I, Littlewood K, Newman J, Boubriak I, Suggitt D, Haydock H, Bennett S, Woodyatt W, Hughes K, Bell J, Coughlan T, van Welsenes D, Kamal M, Cooper C, Tunstall S, Ronan N, Cutts R, Dare T, Yim YTN, Whittley S, Ricamara M, Hamal S, Adams K, Baker H, Driver K, Turner N, Rawlins T, Roy S, Merida-Morillas M, Sakagami Y, Andrews A, Goncalves cordeiro L, Stokes M, Ambihapathy W, Spencer J, Parungao N, Berry L, Cullinane J, Presland L, Ross-Russell A, Warren S, Baker J, Oliver A, Buadi A, Lee K, Haskell L, Romani R, Bentley I, Whitbred T, Fowler S, Gavin J, Magee A, Watson T, Nightingale K, Marius P, Summerton E, Locke E, Honey T, Lingwood A, de la Haye A, Elliott RS, Underwood K, King M, Davies-Dear S, Horsfall E, Chalwin O, Burton H, Edwards CJ, Welham B, Garrahy S, Hall F, Ladikou E, Mullan D, Hansen D, Campbell M, Dos Santos F, Habash-Bailey H, Lakeman N, Branney D, Vamplew L, Hogan A, Frankham J, Wiselka M, Vail D, Wenn V, Renals V, Ellis K, Lewis-Taylor J, Magan J, Hardy A, Appleby K. 2021. Safety and immunogenicity of seven COVID-19 vaccines as a third dose (booster) following two doses of ChAdOx1 nCov-19 or BNT162b2 in the UK (COV-BOOST): a blinded, multicentre, randomised, controlled, phase 2 trial. The Lancet 398:2258–2276.
Pajon R, Doria-Rose NA, Shen X, Schmidt SD, O’Dell S, McDanal C, Feng W, Tong J, Eaton A, Maglinao M, Tang H, Manning KE, Edara V-V, Lai L, Ellis M, Moore KM, Floyd K, Foster SL, Posavad CM, Atmar RL, Lyke KE, Zhou T, Wang L, Zhang Y, Gaudinski MR, Black WP, Gordon I, Guech M, Ledgerwood JE, Misasi JN, Widge A, Sullivan NJ, Roberts PC, Beigel JH, Korber B, Baden LR, El Sahly H, Chalkias S, Zhou H, Feng J, Girard B, Das R, Aunins A, Edwards DK, Suthar MS, Mascola JR, Montefiori DC. 2022. SARS-CoV-2 Omicron Variant Neutralization after mRNA-1273 Booster Vaccination. N Engl J Med 386:1088–1091.
Planas D, Bruel T, Grzelak L, Guivel-Benhassine F, Staropoli I, Porrot F, Planchais C, Buchrieser J, Rajah MM, Bishop E, Albert M, Donati F, Prot M, Behillil S, Enouf V, Maquart M, Smati-Lafarge M, Varon E, Schortgen F, Yahyaoui L, Gonzalez M, De Sèze J, Péré H, Veyer D, Sève A, Simon-Lorière E, Fafi-Kremer S, Stefic K, Mouquet H, Hocqueloux L, van der Werf S, Prazuck T, Schwartz O. 2021. Sensitivity of infectious SARS-CoV-2 B.1.1.7 and B.1.351 variants to neutralizing antibodies. Nat Med 27:917–924.
1669.
Wang P, Nair MS, Liu L, Iketani S, Luo Y, Guo Y, Wang M, Yu J, Zhang B, Kwong PD, Graham BS, Mascola JR, Chang JY, Yin MT, Sobieszczyk M, Kyratsous CA, Shapiro L, Sheng Z, Huang Y, Ho DD. 2021. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 593:130–135.
Kuhlmann C, Mayer CK, Claassen M, Maponga TG, Sutherland AD, Suliman T, Shaw M, Preiser W. 2021. Breakthrough Infections with SARS-CoV-2 Omicron Variant Despite Booster Dose of mRNA Vaccine. SSRN Journal https://doi.org/10.2139/ssrn.3981711.
1673.
Self WH, Tenforde MW, Rhoads JP, Gaglani M, Ginde AA, Douin DJ, Olson SM, Talbot HK, Casey JD, Mohr NM, Zepeski A, McNeal T, Ghamande S, Gibbs KW, Files DC, Hager DN, Shehu A, Prekker ME, Erickson HL, Gong MN, Mohamed A, Henning DJ, Steingrub JS, Peltan ID, Brown SM, Martin ET, Monto AS, Khan A, Hough CL, Busse LW, ten Lohuis CC, Duggal A, Wilson JG, Gordon AJ, Qadir N, Chang SY, Mallow C, Rivas C, Babcock HM, Kwon JH, Exline MC, Halasa N, Chappell JD, Lauring AS, Grijalva CG, Rice TW, Jones ID, Stubblefield WB, Baughman A, Womack KN, Lindsell CJ, Hart KW, Zhu Y, Mills L, Lester SN, Stumpf MM, Naioti EA, Kobayashi M, Verani JR, Thornburg NJ, Patel MM, Calhoun N, Murthy K, Herrick J, McKillop A, Hoffman E, Zayed M, Smith M, Seattle N, Ettlinger J, Priest E, Thomas J, Arroliga A, Beeram M, Kindle R, Kozikowski L-A, De Souza L, Ouellette S, Thornton-Thompson S, Mehkri O, Ashok K, Gole S, King A, Poynter B, Stanley N, Hendrickson A, Maruggi E, Scharber T, Jorgensen J, Bowers R, King J, Aston V, Armbruster B, Rothman RE, Nair R, Chen J-TT, Karow S, Robart E, Maldonado PN, Khan M, So P, Levitt J, Perez C, Visweswaran A, Roque J, Rivera A, Angeles L, Frankel T, Angeles L, Goff J, Huynh D, Howell M, Friedel J, Tozier M, Driver C, Carricato M, Foster A, Nassar P, Stout L, Sibenaller Z, Walter A, Mares J, Olson L, Clinansmith B, Rivas C, Gershengorn H, McSpadden E, Truscon R, Kaniclides A, Thomas L, Bielak R, Valvano WD, Fong R, Fitzsimmons WJ, Blair C, Valesano AL, Gilbert J, Crider CD, Steinbock KA, Paulson TC, Anderson LA, Kampe C, Johnson J, McHenry R, Blair M, Conway D, LaRose M, Landreth L, Hicks M, Parks L, Bongu J, McDonald D, Cass C, Seiler S, Park D, Hink T, Wallace M, Burnham C-A, Arter OG. 2021. Comparative Effectiveness of Moderna, Pfizer-BioNTech, and Janssen (Johnson & Johnson) Vaccines in Preventing COVID-19 Hospitalizations Among Adults Without Immunocompromising Conditions — United States, March–August 2021. MMWR Morb Mortal Wkly Rep 70:1337–1343.
Pagotto V, Ferloni A, Soriano MM, Díaz M, Golde MB, González MI, Asprea V, Staneloni I, Vidal G, Silveira M, Zingoni P, Aliperti V, Michelangelo H, Figar S. 2021. ACTIVE SURVEILLANCE OF THE SPUTNIK V VACCINE IN HEALTH WORKERS. Cold Spring Harbor Laboratory.
He X, Chandrashekar A, Zahn R, Wegmann F, Yu J, Mercado NB, McMahan K, Martinot AJ, Piedra-Mora C, Beecy S, Ducat S, Chamanza R, Huber SR, van der Fits L, Borducchi EN, Lifton M, Liu J, Nampanya F, Patel S, Peter L, Tostanoski LH, Pessaint L, Van Ry A, Finneyfrock B, Velasco J, Teow E, Brown R, Cook A, Andersen H, Lewis MG, Schuitemaker H, Barouch DH. 2021. Low-Dose Ad26.COV2.S Protection Against SARS-CoV-2 Challenge in Rhesus Macaques. Cold Spring Harbor Laboratory.
Stephenson KE, Le Gars M, Sadoff J, de Groot AM, Heerwegh D, Truyers C, Atyeo C, Loos C, Chandrashekar A, McMahan K, Tostanoski LH, Yu J, Gebre MS, Jacob-Dolan C, Li Z, Patel S, Peter L, Liu J, Borducchi EN, Nkolola JP, Souza M, Tan CS, Zash R, Julg B, Nathavitharana RR, Shapiro RL, Azim AA, Alonso CD, Jaegle K, Ansel JL, Kanjilal DG, Guiney CJ, Bradshaw C, Tyler A, Makoni T, Yanosick KE, Seaman MS, Lauffenburger DA, Alter G, Struyf F, Douoguih M, Van Hoof J, Schuitemaker H, Barouch DH. 2021. Immunogenicity of the Ad26.COV2.S Vaccine for COVID-19. JAMA 325:1535.
1692.
McMahan K, Yu J, Mercado NB, Loos C, Tostanoski LH, Chandrashekar A, Liu J, Peter L, Atyeo C, Zhu A, Bondzie EA, Dagotto G, Gebre MS, Jacob-Dolan C, Li Z, Nampanya F, Patel S, Pessaint L, Van Ry A, Blade K, Yalley-Ogunro J, Cabus M, Brown R, Cook A, Teow E, Andersen H, Lewis MG, Lauffenburger DA, Alter G, Barouch DH. 2020. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature 590:630–634.
FDA/CBER. 2020.
Vaccines and Related Biological Products Advisory Committee December 17, 2020 Meeting Briefing Document - FDA
. https://www.fda.gov/media/144434/download.
Mulligan MJ, Lyke KE, Kitchin N, Absalon J, Gurtman A, Lockhart S, Neuzil K, Raabe V, Bailey R, Swanson KA, Li P, Koury K, Kalina W, Cooper D, Fontes-Garfias C, Shi P-Y, Türeci Ö, Tompkins KR, Walsh EE, Frenck R, Falsey AR, Dormitzer PR, Gruber WC, Şahin U, Jansen KU. 2020. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 586:589–593.
1706.
Sahin U, Muik A, Derhovanessian E, Vogler I, Kranz LM, Vormehr M, Baum A, Pascal K, Quandt J, Maurus D, Brachtendorf S, Lörks V, Sikorski J, Hilker R, Becker D, Eller A-K, Grützner J, Boesler C, Rosenbaum C, Kühnle M-C, Luxemburger U, Kemmer-Brück A, Langer D, Bexon M, Bolte S, Karikó K, Palanche T, Fischer B, Schultz A, Shi P-Y, Fontes-Garfias C, Perez JL, Swanson KA, Loschko J, Scully IL, Cutler M, Kalina W, Kyratsous CA, Cooper D, Dormitzer PR, Jansen KU, Türeci Ö. 2020. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 586:594–599.
Castillo-Olivares J, Wells DA, Ferrari M, Chan A, Smith P, Nadesalingam A, Paloniemi M, Carnell G, Ohlendorf L, Cantoni D, Mayora-Neto M, Palmer P, Tonks P, Temperton N, Wagner R, Neckermann P, Peterhoff D, Doffinger R, Kempster S, Otter A, Semper A, Brooks T, Page M, Albecka A, James LC, Briggs J, Schwaeble W, Baxendale H, Heeney J. 2021. Towards Internationally standardised humoral Immune Correlates of Protection from SARS-CoV-2 infection and COVID-19 disease. Cold Spring Harbor Laboratory.
James PT, Ali Z, Armitage AE, Bonell A, Cerami C, Drakesmith H, Jobe M, Jones KS, Liew Z, Moore SE, Morales-Berstein F, Nabwera HM, Nadjm B, Pasricha S-R, Scheelbeek P, Silver MJ, Teh MR, Prentice AM. 2020. Could nutrition modulate COVID-19 susceptibility and severity of disease? A systematic review. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.10.19.20214395.
COVID-19 temporarily bolsters European interest in supplements. nutritioninsightcom/. https://ni.cnsmedia.com/a/EHHJsDOG2oc=. Retrieved 8 February 2021.
Dietary Supplement Health and Education Act of 1994. National Institutes of Health Office of Dietary Supplements. https://ods.od.nih.gov/About/DSHEA_Wording.aspx. Retrieved 25 October 1994.
Bikdeli B, Madhavan MV, Jimenez D, Chuich T, Dreyfus I, Driggin E, Nigoghossian CD, Ageno W, Madjid M, Guo Y, Tang LV, Hu Y, Giri J, Cushman M, Quéré I, Dimakakos EP, Gibson CM, Lippi G, Favaloro EJ, Fareed J, Caprini JA, Tafur AJ, Burton JR, Francese DP, Wang EY, Falanga A, McLintock C, Hunt BJ, Spyropoulos AC, Barnes GD, Eikelboom JW, Weinberg I, Schulman S, Carrier M, Piazza G, Beckman JA, Steg PG, Stone GW, Rosenkranz S, Goldhaber SZ, Parikh SA, Monreal M, Krumholz HM, Konstantinides SV, Weitz JI, Lip GYH. 2020. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up. Journal of the American College of Cardiology 75:2950–2973.
An Investigation on the Effects of Icosapent Ethyl (VascepaTM) on Inflammatory Biomarkers in Individuals With COVID-19 - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04412018. Retrieved 8 February 2021.
A Study of Hydroxychloroquine and Zinc in the Prevention of COVID-19 Infection in Military Healthcare Workers - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04377646. Retrieved 8 February 2021.
Cavalcanti AB, Zampieri FG, Rosa RG, Azevedo LCP, Veiga VC, Avezum A, Damiani LP, Marcadenti A, Kawano-Dourado L, Lisboa T, Junqueira DLM, de Barros e Silva PGM, Tramujas L, Abreu-Silva EO, Laranjeira LN, Soares AT, Echenique LS, Pereira AJ, Freitas FGR, Gebara OCE, Dantas VCS, Furtado RHM, Milan EP, Golin NA, Cardoso FF, Maia IS, Hoffmann Filho CR, Kormann APM, Amazonas RB, Bocchi de Oliveira MF, Serpa-Neto A, Falavigna M, Lopes RD, Machado FR, Berwanger O. 2020. Hydroxychloroquine with or without Azithromycin in Mild-to-Moderate Covid-19. New England Journal of Medicine 383:2041–2052.
Narayanan N, Nair DT. 2020. Vitamin B12 May Inhibit RNA-Dependent-RNA Polymerase Activity of nsp12 from the COVID-19 Virus. Preprints https://doi.org/10.20944/preprints202003.0347.v1.
Hemilä H, Chalker E. 2013. Vitamin C for preventing and treating the common cold. Cochrane Database of Systematic Reviews https://doi.org/10.1002/14651858.cd000980.pub4.
Zhang J, Rao X, Li Y, Zhu Y, Liu F, Guo G, Luo G, Meng Z, Backer DD, Xiang H, Peng Z-Y. 2020. Pilot Trial of High-dose vitamin C in critically ill COVID-19 patients. Research Square https://doi.org/10.21203/rs.3.rs-52778/v2.
1873.
Zhang J, Rao X, Li Y, Zhu Y, Liu F, Guo G, Luo G, Meng Z, Backer DD, Xiang H, Peng Z-Y. 2020. High-dose vitamin C infusion for the treatment of critically ill COVID-19. Research Square https://doi.org/10.21203/rs.3.rs-52778/v1.
1874.
Panel on Dietary Antioxidants and Related Compounds, Subcommittee on Upper Reference Levels of Nutrients, Subcommittee on Interpretation and Uses of Dietary Reference Intakes, Standing Committee on the Scientific Evaluation of Dietary Reference Intakes, Food and Nutrition Board, Institute of Medicine. 2000. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. The National Academies Press. https://doi.org/ghtvqx.
De Smet D, De Smet K, Herroelen P, Gryspeerdt S, Martens GA. 2020. Vitamin D deficiency as risk factor for severe COVID-19: a convergence of two pandemics. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.05.01.20079376.
Hernández JL, Nan D, Fernandez-Ayala M, García-Unzueta M, Hernández-Hernández MA, López-Hoyos M, Muñoz-Cacho P, Olmos JM, Gutiérrez-Cuadra M, Ruiz-Cubillán JJ, Crespo J, Martínez-Taboada VM. 2020. Vitamin D Status in Hospitalized Patients with SARS-CoV-2 Infection. The Journal of Clinical Endocrinology & Metabolism dgaa733.
Carpagnano GE, Di Lecce V, Quaranta VN, Zito A, Buonamico E, Capozza E, Palumbo A, Di Gioia G, Valerio VN, Resta O. 2020. Vitamin D deficiency as a predictor of poor prognosis in patients with acute respiratory failure due to COVID-19. Journal of Endocrinological Investigation https://doi.org/10.1007/s40618-020-01370-x.
Hastie CE, Mackay DF, Ho F, Celis-Morales CA, Katikireddi SV, Niedzwiedz CL, Jani BD, Welsh P, Mair FS, Gray SR, O’Donnell CA, Gill JMR, Sattar N, Pell JP. 2020. Vitamin D concentrations and COVID-19 infection in UK Biobank. Diabetes & Metabolic Syndrome: Clinical Research & Reviews 14:561–565.
1903.
Hastie CE, Pell JP, Sattar N. 2020. Vitamin D and COVID-19 infection and mortality in UK Biobank. European Journal of Nutrition https://doi.org/10.1007/s00394-020-02372-4.
Martín Giménez VM, Inserra F, Ferder L, García J, Manucha W. 2020. Vitamin D deficiency in African Americans is associated with a high risk of severe disease and mortality by SARS-CoV-2. Journal of Human Hypertension https://doi.org/10.1038/s41371-020-00398-z.
Jungreis I, Kellis M. 2020. Mathematical analysis of Córdoba calcifediol trial suggests strong role for Vitamin D in reducing ICU admissions of hospitalized COVID-19 patients. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.11.08.20222638.
COvid-19 and Vitamin D Supplementation: a Multicenter Randomized Controlled Trial of High Dose Versus Standard Dose Vitamin D3 in High-risk COVID-19 Patients (CoVitTrial) - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04344041. Retrieved 8 February 2021.
Parva NR, Tadepalli S, Singh P, Qian A, Joshi R, Kandala H, Nookala VK, Cheriyath P. 2018. Prevalence of Vitamin D Deficiency and Associated Risk Factors in the US Population (2011-2012). Cureus https://doi.org/10.7759/cureus.2741.
Bermudez-Brito M, Plaza-Díaz J, Muñoz-Quezada S, Gómez-Llorente C, Gil A. 2012. Probiotic Mechanisms of Action. Annals of Nutrition and Metabolism 61:160–174.
Zhang H, Kang Z, Gong H, Xu D, Wang J, Li Z, Cui X, Xiao J, Meng T, Zhou W, Liu J, Xu H. 2020. The digestive system is a potential route of 2019-nCov infection: a bioinformatics analysis based on single-cell transcriptomes. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.01.30.927806.
Freedman SB, Williamson-Urquhart S, Farion KJ, Gouin S, Willan AR, Poonai N, Hurley K, Sherman PM, Finkelstein Y, Lee BE, Pang X-L, Chui L, Schnadower D, Xie J, Gorelick M, Schuh S. 2018. Multicenter Trial of a Combination Probiotic for Children with Gastroenteritis. New England Journal of Medicine 379:2015–2026.
Cohen J. 2020. Update: Here’s what is known about Trump’s COVID-19 treatment. Science https://doi.org/10.1126/science.abf0974.
1986.
Louca P, Murray B, Klaser K, Graham MS, Mazidi M, Leeming ER, Thompson E, Bowyer R, Drew DA, Nguyen LH, Merino J, Gomez M, Mompeo O, Costeira R, Sudre CH, Gibson R, Steves CJ, Wolf J, Franks PW, Ourselin S, Chan AT, Berry SE, Valdes AM, Calder PC, Spector TD, Menni C. 2020. Dietary supplements during the COVID-19 pandemic: insights from 1.4M users of the COVID Symptom Study app - a longitudinal app-based community survey. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.11.27.20239087.
Ahmed M, Advani S, Moreira A, Zoretic S, Martinez J, Chorath K, Acosta S, Naqvi R, Burmeister-Morton F, Burmeister F, Tarriela A, Petershack M, Evans M, Hoang A, Rajasekaran K, Ahuja S, Moreira A. 2020. Multisystem inflammatory syndrome in children: A systematic review. EClinicalMedicine 26:100527.
Park SW, Sun K, Viboud C, Grenfell BT, Dushoff J. 2020. Potential roles of social distancing in mitigating the spread of coronavirus disease 2019 (COVID-19) in South Korea. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.27.20045815.
Shahid Z, Kalayanamitra R, McClafferty B, Kepko D, Ramgobin D, Patel R, Aggarwal CS, Vunnam R, Sahu N, Bhatt D, Jones K, Golamari R, Jain R. 2020. ‐19 and Older Adults: What We Know. Journal of the American Geriatrics Society 68:926–929.
Zhu L, She Z-G, Cheng X, Qin J-J, Zhang X-J, Cai J, Lei F, Wang H, Xie J, Wang W, Li H, Zhang P, Song X, Chen X, Xiang M, Zhang C, Bai L, Xiang D, Chen M-M, Liu Y, Yan Y, Liu M, Mao W, Zou J, Liu L, Chen G, Luo P, Xiao B, Zhang C, Zhang Z, Lu Z, Wang J, Lu H, Xia X, Wang D, Liao X, Peng G, Ye P, Yang J, Yuan Y, Huang X, Guo J, Zhang B-H, Li H. 2020. Association of Blood Glucose Control and Outcomes in Patients with COVID-19 and Pre-existing Type 2 Diabetes. Cell Metabolism 31:1068–1077.e3.
2016.
Wu Z, Tang Y, Cheng Q. 2020. Diabetes increases the mortality of patients with COVID-19: a meta-analysis. Acta Diabetologica https://doi.org/10.1007/s00592-020-01546-0.
2017.
Li J, Wang X, Chen J, Zuo X, Zhang H, Deng A. 2020. COVID-19 infection may cause ketosis and ketoacidosis. Diabetes, Obesity and Metabolism https://doi.org/10.1111/dom.14057.
Docherty A, Harrison E, Green C, Hardwick H, Pius R, Norman L, Holden K, Read J, Dondelinger F, Carson G, Merson L, Lee J, Plotkin D, Sigfrid L, Halpin S, Jackson C, Gamble C, Horby P, Nguyen-Van-Tam J, Dunning J, Openshaw P, Baillie J, Semple M, ISARIC4C Investigators. 2020. Features of 16,749 hospitalised UK patients with COVID-19 using the ISARIC WHO Clinical Characterisation Protocol. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.04.23.20076042.
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche J-D, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent J-L, Angus DC. 2016. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315:801.
Uyoga S, M.O. Adetifa I, Karanja HK, Nyagwange J, Tuju J, Wanjiku P, Aman R, Mwangangi M, Amoth P, Kasera K, Ng’ang’a W, Rombo C, Yegon C, Kithi K, Odhiambo E, Rotich T, Orgut I, Kihara S, Otiende M, Bottomley C, N. Mupe Z, W. Kagucia E, E Gallagher K, Etyang A, Voller S, N. Gitonga J, Mugo D, N. Agoti C, Otieno E, Ndwiga L, Lambe T, Wright D, Barasa E, Tsofa B, Bejon P, I. Ochola-Oyier L, Agweyu A, Scott JAG, Warimwe GM. 2020. Seroprevalence of anti-SARS-CoV-2 IgG antibodies in Kenyan blood donors. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.07.27.20162693.
2042.
Chibwana MG, Jere KC, Kamn’gona R, Mandolo J, Katunga-Phiri V, Tembo D, Mitole N, Musasa S, Sichone S, Lakudzala A, Sibale L, Matambo P, Kadwala I, Byrne RL, Mbewe A, Morton B, Phiri C, Mallewa J, Mwandumba HC, Adams ER, Gordon SB, Jambo KC. 2020. High SARS-CoV-2 seroprevalence in Health Care Workers but relatively low numbers of deaths in urban Malawi. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.07.30.20164970.
Dasgupta N, Funk MJ, Lazard A, White BE, Marshall SW. 2020. Quantifying the social distancing privilege gap: a longitudinal study of smartphone movement. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.05.03.20084624.
Żukiewicz-Sobczak W, Wróblewska P, Zwoliński J, Chmielewska-Badora J, Adamczuk P, Krasowska E, Zagórski J, Oniszczuk A, Piątek J, Silny W. 2014. Obesity and poverty paradox in developed countries. Annals of Agricultural and Environmental Medicine 21:590–594.
Miller GE, Blackwell E. 2016. Turning Up the Heat. Current Directions in Psychological Science 15:269–272.
2076.
Sapolsky R. 2005. Sick of Poverty. Scientific American 293:92–99.
2077.
Wu X, Nethery RC, Sabath MB, Braun D, Dominici F. 2020. Exposure to air pollution and COVID-19 mortality in the United States: A nationwide cross-sectional study. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.04.05.20054502.
2078.
Burnett R, Chen H, Szyszkowicz M, Fann N, Hubbell B, Pope CA, Apte JS, Brauer M, Cohen A, Weichenthal S, Coggins J, Di Q, Brunekreef B, Frostad J, Lim SS, Kan H, Walker KD, Thurston GD, Hayes RB, Lim CC, Turner MC, Jerrett M, Krewski D, Gapstur SM, Diver WR, Ostro B, Goldberg D, Crouse DL, Martin RV, Peters P, Pinault L, Tjepkema M, van Donkelaar A, Villeneuve PJ, Miller AB, Yin P, Zhou M, Wang L, Janssen NAH, Marra M, Atkinson RW, Tsang H, Quoc Thach T, Cannon JB, Allen RT, Hart JE, Laden F, Cesaroni G, Forastiere F, Weinmayr G, Jaensch A, Nagel G, Concin H, Spadaro JV. 2018. Global estimates of mortality associated with long-term exposure to outdoor fine particulate matter. Proceedings of the National Academy of Sciences 115:9592–9597.
Shah M, Sachdeva M, Dodiuk-Gad RP. 2020. COVID-19 and racial disparities. Journal of the American Academy of Dermatology 83:e35.
2088.
FAQs for COVID-19 Claims Reimbursement to Health Care Providers and Facilities for Testing, Treatment and Vaccine Administration. Official web site of the US Health Resources & Services Administration. Text. https://www.hrsa.gov/coviduninsuredclaim/frequently-asked-questions. Retrieved 8 February 2021.
Raffensperger J, Brauner M, Briggs R. 2020. Planning Hospital Needs for Ventilators and Respiratory Therapists in the COVID-19 Crisis. Rand Corporation. https://doi.org/ghfprp.
Wang LL, Lo K, Chandrasekhar Y, Reas R, Yang J, Burdick D, Eide D, Funk K, Katsis Y, Kinney R, Li Y, Liu Z, Merrill W, Mooney P, Murdick D, Rishi D, Sheehan J, Shen Z, Stilson B, Wade A, Wang K, Wang NXR, Wilhelm C, Xie B, Raymond D, Weld DS, Etzioni O, Kohlmeier S. 2020. CORD-19: The COVID-19 Open Research Dataset. 2004.10706arXiv. arXiv.
Riegelman RK. 2013. Studying a study & testing a test: reading evidence-based health research6th ed. Wolters Kluwer/Lippincott Williams & Wilkins Heath, Philadelphia.
Tian X, Li C, Huang A, Xia S, Lu S, Shi Z, Lu L, Jiang S, Yang Z, Wu Y, Ying T. 2020. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.01.28.923011.
2163.
He X, Zhang L, Ran Q, Wang J, Xiong A, Wu D, Chen F, Li G. 2020. Integrative Bioinformatics Analysis Provides Insight into the Molecular Mechanisms of 2019-nCoV. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.03.20020206.
2164.
Liang W, Feng Z, Rao S, Xiao C, Lin Z-X, Zhang Q, Wei Q. 2020. Diarrhea may be underestimated: a missing link in 2019 novel coronavirus. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.03.20020289.
2165.
Chai X, Hu L, Zhang Y, Han W, Lu Z, Ke A, Zhou J, Shi G, Fang N, Fan J, Cai J, Fan J, Lan F. 2020. Specific ACE2 Expression in Cholangiocytes May Cause Liver Damage After 2019-nCoV Infection. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.03.931766.
2166.
Zhao B, Ni C, Gao R, Wang Y, Yang L, Wei J, Lv T, Liang J, Zhang Q, Xu W, Xie Y, Wang X, Yuan Z, Liang J, Zhang R, Lin X. 2020. Recapitulation of SARS-CoV-2 Infection and Cholangiocyte Damage with Human Liver Organoids. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.16.990317.
2167.
Wang J, Zhao S, Liu M, Zhao Z, Xu Y, Wang P, Lin M, Xu Y, Huang B, Zuo X, Chen Z, Bai F, Cui J, Lew AM, Zhao J, Zhang Y, Luo H-B, Zhang Y. 2020. ACE2 expression by colonic epithelial cells is associated with viral infection, immunity and energy metabolism. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.05.20020545.
2168.
Bao L, Deng W, Huang B, Gao H, Liu J, Ren L, Wei Q, Yu P, Xu Y, Qi F, Qu Y, Li F, Lv Q, Wang W, Xue J, Gong S, Liu M, Wang G, Wang S, Song Z, Zhao L, Liu P, Zhao L, Ye F, Wang H, Zhou W, Zhu N, Zhen W, Yu H, Zhang X, Guo L, Chen L, Wang C, Wang Y, Wang X, Xiao Y, Sun Q, Liu H, Zhu F, Ma C, Yan L, Yang M, Han J, Xu W, Tan W, Peng X, Jin Q, Wu G, Qin C. 2020. The Pathogenicity of SARS-CoV-2 in hACE2 Transgenic Mice. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.07.939389.
2169.
Li Z, Wu M, Yao J, Guo J, Liao X, Song S, Li J, Duan G, Zhou Y, Wu X, Zhou Z, Wang T, Hu M, Chen X, Fu Y, Lei C, Dong H, Xu C, Hu Y, Han M, Zhou Y, Jia H, Chen X, Yan J. 2020. Caution on Kidney Dysfunctions of COVID-19 Patients. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.08.20021212.
Lin W, Hu L, Zhang Y, Ooi JD, Meng T, Jin P, Ding X, Peng L, Song L, Xiao Z, Ao X, Xiao X, Zhou Q, Xiao P, Fan J, Zhong Y. 2020. Single-cell Analysis of ACE2 Expression in Human Kidneys and Bladders Reveals a Potential Route of 2019-nCoV Infection. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.08.939892.
2172.
Zhu J, Kim J, Xiao X, Wang Y, Luo D, Jiang S, Chen R, Xu L, Zhang H, Moise L, Gutierrez AH, De Groot AS, Xiao G, Schoggins JW, Zhan X, Wang T, Xie Y. 2020. The immune vulnerability landscape of the 2019 Novel Coronavirus, SARS-CoV-2. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.08.939553.
Liu J, Liu Y, Xiang P, Pu L, Xiong H, Li C, Zhang M, Tan J, Xu Y, Song R, Song M, Wang L, Zhang W, Han B, Yang L, Wang X, Zhou G, Zhang T, Li B, Wang Y, Chen Z, Wang X. 2020. Neutrophil-to-Lymphocyte Ratio Predicts Severe Illness Patients with 2019 Novel Coronavirus in the Early Stage. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.10.20021584.
Wan S, Yi Q, Fan S, Lv J, Zhang X, Guo L, Lang C, Xiao Q, Xiao K, Yi Z, Qiang M, Xiang J, Zhang B, Chen Y. 2020. Characteristics of lymphocyte subsets and cytokines in peripheral blood of 123 hospitalized patients with 2019 novel coronavirus pneumonia (NCP). Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.10.20021832.
2177.
Liu J, Li S, Liu J, Liang B, Wang X, Li W, Wang H, Tong Q, Yi J, Zhao L, Xiong L, Guo C, Tian J, Luo J, Yao J, Pang R, Shen H, Peng C, Liu T, Zhang Q, Wu J, Xu L, Lu S, Wang B, Weng Z, Han C, Zhu H, Zhou R, Zhou H, Chen X, Ye P, Zhu B, He S, He Y, Jie S, Wei P, Zhang J, Lu Y, Wang W, Zhang L, Li L, Zhou F, Wang J, Dittmer U, Lu M, Hu Y, Yang D, Zheng X. 2020. Longitudinal Characteristics of Lymphocyte Responses and Cytokine Profiles in the Peripheral Blood of SARS-CoV-2 Infected Patients. SSRN Electronic Journal https://doi.org/10.2139/ssrn.3539682.
2178.
Li J, Li S, Cai Y, Liu Q, Li X, Zeng Z, Chu Y, Zhu F, Zeng F. 2020. Epidemiological and Clinical Characteristics of 17 Hospitalized Patients with 2019 Novel Coronavirus Infections Outside Wuhan, China. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.11.20022053.
2179.
Fan C, Li K, Ding Y, Lu W, Wang J. 2020. ACE2 Expression in Kidney and Testis May Cause Kidney and Testis Damage After 2019-nCoV Infection. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.12.20022418.
2180.
Zhou Y, Fu B, Zheng X, Wang D, Zhao C, qi Y, Sun R, Tian Z, Xu X, Wei H. 2020. Aberrant pathogenic GM-CSF <sup>+</sup> T cells and inflammatory CD14 <sup>+</sup> CD16 <sup>+</sup> monocytes in severe pulmonary syndrome patients of a new coronavirus. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.12.945576.
2181.
Qian K, Deng Y, Tai Y-H, Peng J, Peng H, Jiang L-H. 2020. Clinical Characteristics of 2019 Novel Infected Coronavirus Pneumonia: A Systemic Review and Meta-analysis. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.14.20021535.
2182.
Liu J, Li S, Liu J, Liang B, Wang X, Wang H, Li W, Tong Q, Yi J, Zhao L, Xiong L, Guo C, Tian J, Luo J, Yao J, Pang R, Shen H, Peng C, Liu T, Zhang Q, Wu J, Xu L, Lu S, Wang B, Weng Z, Han C, Zhu H, Zhou R, Zhou H, Chen X, Ye P, Zhu B, He S, He Y, Jie S, Wei P, Zhang J, Lu Y, Wang W, Zhang L, Li L, Zhou F, Wang J, Dittmer U, Lu M, Hu Y, Yang D, Zheng X. 2020. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.16.20023671.
2183.
Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, Wang T, Zhang X, Chen H, Yu H, Zhang X, Zhang M, Wu S, Song J, Chen T, Han M, Li S, Luo X, Zhao J, Ning Q. 2020. Clinical and immunologic features in severe and moderate forms of Coronavirus Disease 2019. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.16.20023903.
2184.
Chen Y, Wei Q, Li R, Gao H, Zhu H, Deng W, Bao L, Tong W, Cong Z, Jiang H, Qin C. 2020. Protection of Rhesus Macaque from SARS-Coronavirus challenge by recombinant adenovirus vaccine. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.17.951939.
2185.
Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, Chen L, Li M, Liu Y, Wang G, Yuan Z, Feng Z, Wu Y, Chen Y. 2020. Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.18.20024364.
2186.
Li X, Wang L, Yan S, Yang F, Xiang L, Zhu J, Shen B, Gong Z. 2020. Clinical characteristics of 25 death cases with COVID-19: a retrospective review of medical records in a single medical center, Wuhan, China. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.19.20025239.
2187.
Wang L, Li X, Chen H, Yan S, Li Y, Li D, Gong Z. 2020. SARS-CoV-2 infection does not significantly cause acute renal injury: an analysis of 116 hospitalized patients with COVID-19 in a single hospital, Wuhan, China. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.19.20025288.
Guan W, Ni Z, Hu Y, Liang W, Ou C, He J, Liu L, Shan H, Lei C, Hui DSC, Du B, Li L, Zeng G, Yuen K-Y, Chen R, Tang C, Wang T, Chen P, Xiang J, Li S, Wang J, Liang Z, Peng Y, Wei L, Liu Y, Hu Y, Peng P, Wang J, Liu J, Chen Z, Li G, Zheng Z, Qiu S, Luo J, Ye C, Zhu S, Zhong N. 2020. Clinical characteristics of 2019 novel coronavirus infection in China. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.06.20020974.
2190.
Fast E, Altman RB, Chen B. 2020. Potential T-cell and B-cell Epitopes of 2019-nCoV. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.19.955484.
2191.
Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. 2020. Structure, function and antigenicity of the SARS-CoV-2 spike glycoprotein. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.19.956581.
2192.
Thevarajan I, Nguyen TH, Koutsakos M, Druce J, Caly L, van de Sandt CE, Jia X, Nicholson S, Catton M, Cowie B, Tong SY, Lewin SR, Kedzierska K. 2020. Breadth of concomitant immune responses underpinning viral clearance and patient recovery in a non-severe case of COVID-19. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.20.20025841.
2193.
Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, Chen L, Li J, Wang X, Wang F, Liu L, Zhang S, Zhang Z. 2020. The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.23.20026690.
Pan Y, Ye G, Zeng X, Liu G, Zeng X, Jiang X, Zhao J, Chen L, Guo S, Deng Q, Hong X, Yang Y, Li Y, Wang X. 2020. Can routine laboratory tests discriminate 2019 novel coronavirus infected pneumonia from other community-acquired pneumonia? Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.25.20024711.
2200.
Gong J, Dong H, Xia Q, Huang Z, Wang D, Zhao Y, Liu W, Tu S, Zhang M, Wang Q, Lu F. 2020. Correlation Analysis Between Disease Severity and Inflammation-related Parameters in Patients with COVID-19 Pneumonia. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.25.20025643.
2201.
Herst C, Burkholz S, Sidney J, Sette A, Harris P, Massey S, Brasel T, Cunha-Neto E, Rosa D, Chao W, Carback R, Hodge T, Wang L, Ciotlos S, Lloyd P, Rubsamen R. 2020. An Effective CTL Peptide Vaccine for Ebola Zaire Based on Survivors’ CD8+ Targeting of a Particular Nucleocapsid Protein Epitope with Potential Implications for COVID-19 Vaccine Design. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.25.963546.
2202.
Li L, Sun T, He Y, Li W, Fan Y, Zhang J. 2020. Epitope-based peptide vaccine design and target site characterization against novel coronavirus disease caused by SARS-CoV-2. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.25.965434.
2203.
Wenjun W, Xiaoqing L, Sipei W, Puyi L, Liyan H, Yimin L, Linling C, Sibei C, Lingbo N, Yongping L, Jianxing H. 2020. The definition and risks of Cytokine Release Syndrome-Like in 11 COVID-19-Infected Pneumonia critically ill patients: Disease Characteristics and Retrospective Analysis. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.26.20026989.
2204.
Huang Y, Yang R, Xu Y, Gong P. 2020. Clinical characteristics of 36 non-survivors with COVID-19 in Wuhan, China. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.27.20029009.
2205.
Li L, Li S, Xu M, Yu P, Zheng S, Duan Z, Liu J, Chen Y, Li J. 2020. Risk factors related to hepatic injury in patients with corona virus disease 2019. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.28.20028514.
2206.
Chen X, Zhao B, Qu Y, Chen Y, Xiong J, Feng Y, Men D, Huang Q, Liu Y, Yang B, Ding J, Li F. 2020. Detectable serum SARS-CoV-2 viral load (RNAaemia) is closely associated with drastically elevated interleukin 6 (IL-6) level in critically ill COVID-19 patients. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.29.20029520.
Tan L, Wang Q, Zhang D, Ding J, Huang Q, Tang Y-Q, Wang Q, Miao H. 2020. Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.01.20029074.
2209.
Liu T, Zhang J, Yang Y, Ma H, Li Z, Zhang J, Cheng J, Zhang X, Zhao Y, Xia Z, Zhang L, Wu G, Yi J. 2020. The potential role of IL-6 in monitoring severe case of coronavirus disease 2019. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.01.20029769.
2210.
Zhao Z, Xie J, Yin M, Yang Y, He H, Jin T, Li W, Zhu X, Xu J, Zhao C, Li L, Li Y, Mengist HM, Zahid A, Yao Z, Ding C, Qi Y, Gao Y, Ma X. 2020. Clinical and Laboratory Profiles of 75 Hospitalized Patients with Novel Coronavirus Disease 2019 in Hefei, China. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.01.20029785.
2211.
Yang Y, Shen C, Li J, Yuan J, Yang M, Wang F, Li G, Li Y, Xing L, Peng L, Wei J, Cao M, Zheng H, Wu W, Zou R, Li D, Xu Z, Wang H, Zhang M, Zhang Z, Liu L, Liu Y. 2020. Exuberant elevation of IP-10, MCP-3 and IL-1ra during SARS-CoV-2 infection is associated with disease severity and fatal outcome. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.02.20029975.
2212.
Zhao J, Yuan Q, Wang H, Liu W, Liao X, Su Y, Wang X, Yuan J, Li T, Li J, Qian S, Hong C, Wang F, Liu Y, Wang Z, He Q, Li Z, He B, Zhang T, Ge S, Liu L, Zhang J, Xia N, Zhang Z. 2020. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.02.20030189.
2213.
Chen X, Ling J, Mo P, Zhang Y, Jiang Q, Ma Z, Cao Q, Hu W, Zou S, Chen L, Yao L, Luo M, Chen T, Deng L, Liang K, Song S, Yang R, Zheng R, Gao S, Gui X, Ke H, Hou W, Lundkvist Å, Xiong Y. 2020. Restoration of leukomonocyte counts is associated with viral clearance in COVID-19 hospitalized patients. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.03.20030437.
Xu Y, Xu Z, Liu X, Cai L, Zheng H, Huang Y, Zhou L, Huang L, Lin Y, Deng L, Li J, Chen S, Liu D, Lin Z, Zhou L, He W, Liu X, Li Y. 2020. Clinical findings in critically ill patients infected with SARS-CoV-2 in Guangdong Province, China: a multi-center, retrospective, observational study. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.03.20030668.
2217.
Feng Y, Qiu M, Liu L, Zou S, Li Y, Luo K, Guo Q, Han N, Sun Y, Wang K, Zhuang X, Zhang S, Chen S, Mo F. 2020. Multi-epitope vaccine design using an immunoinformatics approach for 2019 novel coronavirus (SARS-CoV-2). Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.03.962332.
2218.
Cao M, Zhang D, Wang Y, Lu Y, Zhu X, Li Y, Xue H, Lin Y, Zhang M, Sun Y, Yang Z, Shi J, Wang Y, Zhou C, Dong Y, Peng L, Liu P, Dudek SM, Xiao Z, Lu H. 2020. Clinical Features of Patients Infected with the 2019 Novel Coronavirus (COVID-19) in Shanghai, China. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.04.20030395.
2219.
Zhang J, Liu J, Li N, Liu Y, Ye R, Qin X, Zheng R. 2020. Serological detection of 2019-nCoV respond to the epidemic: A useful complement to nucleic acid testing. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.04.20030916.
2220.
Diao B, Wang C, Wang R, Feng Z, Tan Y, Wang H, Wang C, Liu L, Liu Y, Liu Y, Wang G, Yuan Z, Ren L, Wu Y, Chen Y. 2020. Human Kidney is a Target for Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.04.20031120.
2221.
Song C-Y, Xu J, He J-Q, Lu Y-Q. 2020. COVID-19 early warning score: a multi-parameter screening tool to identify highly suspected patients. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.05.20031906.
2222.
Pfaender S, Mar KB, Michailidis E, Kratzel A, Hirt D, V’kovski P, Fan W, Ebert N, Stalder H, Kleine-Weber H, Hoffmann M, Hoffmann HH, Saeed M, Dijkman R, Steinmann E, Wight-Carter M, Hanners NW, Pöhlmann S, Gallagher T, Todt D, Zimmer G, Rice CM, Schoggins JW, Thiel V. 2020. LY6E impairs coronavirus fusion and confers immune control of viral disease. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.05.979260.
2223.
Liu L, Liu W, Zheng Y, Jiang X, Kou G, Ding J, Wang Q, Huang Q, Ding Y, Ni W, Wu W, Tang S, Tan L, Hu Z, Xu W, Zhang Y, Zhang B, Tang Z, Zhang X, Li H, Rao Z, Jiang H, Ren X, Wang S, Zheng S. 2020. A preliminary study on serological assay for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in 238 admitted hospital patients. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.06.20031856.
2224.
Zheng Z, Monteil VM, Maurer-Stroh S, Yew CW, Leong C, Mohd-Ismail NK, Arularasu SC, Chow VTK, Pin RLT, Mirazimi A, Hong W, Tan Y-J. 2020. Monoclonal antibodies for the S2 subunit of spike of SARS-CoV cross-react with the newly-emerged SARS-CoV-2. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.06.980037.
2225.
Zeng Q, Li Y, Huang G, Wu W, Dong S, Xu Y. 2020. Mortality of COVID-19 is Associated with Cellular Immune Function Compared to Immune Function in Chinese Han Population. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.08.20031229.
2226.
Fan H, Zhang L, Huang B, Zhu M, Zhou Y, Zhang H, Tao X, Cheng S, Yu W, Zhu L, Chen J. 2020. Retrospective Analysis of Clinical Features in 101 Death Cases with COVID-19. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.09.20033068.
2227.
Matsuyama S, Kawase M, Nao N, Shirato K, Ujike M, Kamitani W, Shimojima M, Fukushi S. 2020. The inhaled corticosteroid ciclesonide blocks coronavirus RNA replication by targeting viral NSP15. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.11.987016.
2228.
Zhang B, Zhou X, Zhu C, Feng F, Qiu Y, Feng J, Jia Q, Song Q, Zhu B, Wang J. 2020. Immune phenotyping based on neutrophil-to-lymphocyte ratio and IgG predicts disease severity and outcome for patients with COVID-19. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.12.20035048.
2229.
Bao L, Deng W, Gao H, Xiao C, Liu J, Xue J, Lv Q, Liu J, Yu P, Xu Y, Qi F, Qu Y, Li F, Xiang Z, Yu H, Gong S, Liu M, Wang G, Wang S, Song Z, Liu Y, Zhao W, Han Y, Zhao L, Liu X, Wei Q, Qin C. 2020. Lack of Reinfection in Rhesus Macaques Infected with SARS-CoV-2. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.13.990226.
2230.
Yuan M, Wu NC, Zhu X, Lee C-CD, So RTY, Lv H, Mok CKP, Wilson IA. 2020. A highly conserved cryptic epitope in the receptor-binding domains of SARS-CoV-2 and SARS-CoV. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.13.991570.
2231.
Dong L, Zhou J, Niu C, Wang Q, Pan Y, Sheng S, Wang X, Zhang Y, Yang J, Liu M, Zhao Y, Zhang X, Zhu T, Peng T, Xie J, Gao Y, Wang D, Zhao Y, Dai X, Fang X. 2020. Highly accurate and sensitive diagnostic detection of SARS-CoV-2 by digital PCR. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.14.20036129.
2232.
Wang K, Chen W, Zhou Y-S, Lian J-Q, Zhang Z, Du P, Gong L, Zhang Y, Cui H-Y, Geng J-J, Wang B, Sun X-X, Wang C-F, Yang X, Lin P, Deng Y-Q, Wei D, Yang X-M, Zhu Y-M, Zhang K, Zheng Z-H, Miao J-L, Guo T, Shi Y, Zhang J, Fu L, Wang Q-Y, Bian H, Zhu P, Chen Z-N. 2020. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.14.988345.
Weidle UH, Scheuer W, Eggle D, Klostermann S, Stockinger H. Cancer-related issues of CD147. Cancer Genomics Proteomics 7:157–69.
2236.
Huang L, Shi Y, Gong B, Jiang L, Liu X, Yang J, Tang J, You C, Jiang Q, Long B, Zeng T, Luo M, Zeng F, Zeng F, Wang S, Yang X, Yang Z. 2020. Blood single cell immune profiling reveals the interferon-MAPK pathway mediated adaptive immune response for COVID-19. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.15.20033472.
2237.
Lv H, Wu NC, Tsang OT-Y, Yuan M, Perera RAPM, Leung WS, So RTY, Chun Chan JM, Yip GK, Hong Chik TS, Wang Y, Chung Choi CY, Lin Y, Ng WW, Zhao J, Poon LLM, Malik Peiris JS, Wilson IA, Mok CKP. 2020. Cross-reactive antibody response between SARS-CoV-2 and SARS-CoV infections. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.15.993097.
2238.
Duan K, Liu B, Li C, Zhang H, Yu T, Qu J, Zhou M, Chen L, Meng S, Hu Y, Peng C, Yuan M, Huang J, Wang Z, Yu J, Gao X, Wang D, Yu X, Li L, Zhang J, Wu X, Li B, Xu Y, Chen W, Peng Y, Hu Y, Lin L, Liu X, Huang S, Zhou Z, Zhang L, Wang Y, Zhang Z, Deng K, Xia Z, Gong Q, Zhang W, Zheng X, Liu Y, Yang H, Zhou D, Yu D, Hou J, Shi Z, Saijuan C, Chen Z, Zhang X, Yang X. 2020. The feasibility of convalescent plasma therapy in severe COVID- 19 patients: a pilot study. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.16.20036145.
2239.
Gautret P, Lagier J-C, Parola P, Hoang VT, Meddeb L, Mailhe M, Doudier B, Courjon J, Giordanengo V, Vieira VE, Dupont HT, Honoré S, Colson P, Chabrière E, Scola BL, Rolain J-M, Brouqui P, Raoult D. 2020. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.16.20037135.
Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, Zhang Y, Liu Z, Fritz JM, Marsh R, Husami A, Kissell D, Nortman S, Chaturvedi V, Haines H, Young LR, Mo J, Filipovich AH, Bleesing JJ, Mustillo P, Stephens M, Rueda CM, Chougnet CA, Hoebe K, McElwee J, Hughes JD, Karakoc-Aydiner E, Matthews HF, Price S, Su HC, Rao VK, Lenardo MJ, Jordan MB. 2015. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 349:436–440.
2242.
Procko E. 2020. The sequence of human ACE2 is suboptimal for binding the S spike protein of SARS coronavirus 2. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.16.994236.
2243.
Rockx B, Kuiken T, Herfst S, Bestebroer T, Lamers MM, de Meulder D, van Amerongen G, van den Brand J, Okba NMA, Schipper D, van Run P, Leijten L, Verschoor E, Verstrepen B, Langermans J, Drosten C, van Vlissingen MF, Fouchier R, de Swart R, Koopmans M, Haagmans BL. 2020. Comparative Pathogenesis Of COVID-19, MERS And SARS In A Non-Human Primate Model. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.17.995639.
Aguilar JB, Faust JS, Westafer LM, Gutierrez JB. 2020. Modeling the Impact of Asymptomatic Carriers on COVID-19 Transmission Dynamics During Lockdown. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.18.20037994.
2246.
Long Q, Deng H, Chen J, Hu J, Liu B, Liao P, Lin Y, Yu L, Mo Z, Xu Y, Gong F, Wu G, Zhang X, Chen Y, Li Z, Wang K, Zhang X, Tian W, Niu C, Yang Q, Xiang J, Du H, Liu H, Lang C, Luo X-H, Wu S, Cui X, Zhou Z, Wang J, Xue C, Li X, Wang L, Tang X, Zhang Y, Qiu J, Liu X, Li J, Zhang D, Zhang F, Cai X, Wang D, Hu Y, Ren J, Tang N, Liu P, Li Q, Huang A. 2020. Antibody responses to SARS-CoV-2 in COVID-19 patients: the perspective application of serological tests in clinical practice. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.18.20038018.
2247.
Hu X, An T, Situ B, Hu Y, Ou Z, Li Q, He X, Zhang Y, Tian P, Sun D, Rui Y, Wang Q, Ding D, Zheng L. 2020. Heat inactivation of serum interferes with the immunoanalysis of antibodies to SARS-CoV-2. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.12.20034231.
2248.
Okba NMA, Müller MA, Li W, Wang C, GeurtsvanKessel CH, Corman VM, Lamers MM, Sikkema RS, de Bruin E, Chandler FD, Yazdanpanah Y, Le Hingrat Q, Descamps D, Houhou-Fidouh N, Reusken CBEM, Bosch B-J, Drosten C, Koopmans MPG, Haagmans BL. 2020. SARS-CoV-2 specific antibody responses in COVID-19 patients. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.18.20038059.
2249.
Belhadi D, Peiffer-Smadja N, Lescure F-X, Yazdanpanah Y, Mentré F, Laouénan C. 2020. A brief review of antiviral drugs evaluated in registered clinical trials for COVID-19. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.18.20038190.
2250.
Leung JM, Yang CX, Tam A, Shaipanich T, Hackett T-L, Singhera GK, Dorscheid DR, Sin DD. 2020. ACE-2 Expression in the Small Airway Epithelia of Smokers and COPD Patients: Implications for COVID-19. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.18.20038455.
2251.
Xu Y. 2020. Dynamic profile of severe or critical COVID-19 cases. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.18.20038513.
2252.
Fu H, Xu H, Zhang N, Xu H, Li Z, Chen H, Xu R, Sun R, Wen L, Xie L, Liu H, Zhang K, Fu C, Hou K, Yang Z, Yang M, Guo Y. 2020. Association between Clinical, Laboratory and CT Characteristics and RT-PCR Results in the Follow-up of COVID-19 patients. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.19.20038315.
2253.
Sheahan TP, Sims AC, Zhou S, Graham RL, Hill CS, Leist SR, Schäfer A, Dinnon KH, Montgomery SA, Agostini ML, Pruijssers AJ, Chapell JD, Brown AJ, Bluemling GR, Natchus MG, Saindane M, Kolykhalov AA, Painter G, Harcourt J, Tamin A, Thornburg NJ, Swanstrom R, Denison MR, Baric RS. 2020. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 and multiple endemic, epidemic and bat coronavirus. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.19.997890.
2254.
Jeon S, Ko M, Lee J, Choi I, Byun SY, Park S, Shum D, Kim S. 2020. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.20.999730.
2255.
Munster VJ, Feldmann F, Williamson BN, van Doremalen N, Pérez-Pérez L, Schulz J, Meade-White K, Okumura A, Callison J, Brumbaugh B, Avanzato VA, Rosenke R, Hanley PW, Saturday G, Scott D, Fischer ER, de Wit E. 2020. Respiratory disease and virus shedding in rhesus macaques inoculated with SARS-CoV-2. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.21.001628.
2256.
Deng W, Bao L, Gao H, Xiang Z, Qu Y, Song Z, Gong S, Liu J, Liu J, Yu P, Qi F, Xu Y, Li F, Xiao C, Lv Q, Xue J, Wei Q, Liu M, Wang G, Wang S, Yu H, Liu X, Zhao W, Han Y, Qin C. 2020. Ocular conjunctival inoculation of SARS-CoV-2 can cause mild COVID-19 in Rhesus macaques. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.13.990036.
2257.
Pinto BGG, Oliveira AER, Singh Y, Jimenez L, Gonçalves ANA, Ogava RLT, Creighton R, Schatzmann Peron JP, Nakaya HI. 2020. ACE2 Expression is Increased in the Lungs of Patients with Comorbidities Associated with Severe COVID-19. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.21.20040261.
2258.
Bian H, Zheng Z-H, Wei D, Zhang Z, Kang W-Z, Hao C-Q, Dong K, Kang W, Xia J-L, Miao J-L, Xie R-H, Wang B, Sun X-X, Yang X-M, Lin P, Geng J-J, Wang K, Cui H-Y, Zhang K, Chen X-C, Tang H, Du H, Yao N, Liu S-S, Liu L-N, Zhang Z, Gao Z-W, Nan G, Wang Q-Y, Lian J-Q, Chen Z-N, Zhu P. 2020. Meplazumab treats COVID-19 pneumonia: an open-labelled, concurrent controlled add-on clinical trial. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.21.20040691.
Davidson AD, Williamson MK, Lewis S, Shoemark D, Carroll MW, Heesom K, Zambon M, Ellis J, Lewis PA, Hiscox JA, Matthews DA. 2020. Characterisation of the transcriptome and proteome of SARS-CoV-2 using direct RNA sequencing and tandem mass spectrometry reveals evidence for a cell passage induced in-frame deletion in the spike glycoprotein that removes the furin-like cleavage site. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.22.002204.
Kim D, Lee J-Y, Yang J-S, Kim JW, Kim VN, Chang H. 2020. The architecture of SARS-CoV-2 transcriptome. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.12.988865.
2267.
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, O’Meara MJ, Guo JZ, Swaney DL, Tummino TA, Hüttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Naing ZZC, Zhou Y, Peng S, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Shen W, Shi Y, Zhang Z, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Ramachandran R, Liu X, Rosenthal SB, Calviello L, Venkataramanan S, Lin Y, Wankowicz SA, Bohn M, Trenker R, Young JM, Cavero D, Hiatt J, Roth T, Rathore U, Subramanian A, Noack J, Hubert M, Roesch F, Vallet T, Meyer B, White KM, Miorin L, Agard D, Emerman M, Ruggero D, García-Sastre A, Jura N, Zastrow M von, Taunton J, Schwartz O, Vignuzzi M, d’Enfert C, Mukherjee S, Jacobson M, Malik HS, Fujimori DG, Ideker T, Craik CS, Floor S, Fraser JS, Gross J, Sali A, Kortemme T, Beltrao P, Shokat K, Shoichet BK, Krogan NJ. 2020. A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.22.002386.
2268.
Chen H, Zhang Z, Wang L, Huang Z, Gong F, Li X, Chen Y, Wu JJ. 2020. First Clinical Study Using HCV Protease Inhibitor Danoprevir to Treat Naïve and Experienced COVID-19 Patients. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.22.20034041.
2269.
Seiwert SD, Andrews SW, Jiang Y, Serebryany V, Tan H, Kossen K, Rajagopalan PTR, Misialek S, Stevens SK, Stoycheva A, Hong J, Lim SR, Qin X, Rieger R, Condroski KR, Zhang H, Do MG, Lemieux C, Hingorani GP, Hartley DP, Josey JA, Pan L, Beigelman L, Blatt LM. 2008. Preclinical Characteristics of the Hepatitis C Virus NS3/4A Protease Inhibitor ITMN-191 (R7227). Antimicrobial Agents and Chemotherapy 52:4432–4441.
Nguyen DD, Gao K, Chen J, Wang R, Wei G-W. 2020. Potentially highly potent drugs for 2019-nCoV. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.02.05.936013.
2272.
Lou B, Li T-D, Zheng S-F, Su Y-Y, Li Z-Y, Liu W, Yu F, Ge S-X, Zou Q-D, Yuan Q, Lin S, Hong C-M, Yao X-Y, Zhang X-J, Wu D-H, Zhou G-L, Hou W-H, Li T-T, Zhang Y-L, Zhang S-Y, Fan J, Zhang J, Xia N-S, Chen Y. 2020. Serology characteristics of SARS-CoV-2 infection since the exposure and post symptoms onset. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.23.20041707.
2273.
Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Møller R, Panis M, Sachs D, Albrecht RA, tenOever BR. 2020. SARS-CoV-2 launches a unique transcriptional signature from in vitro, ex vivo, and in vivo systems. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.24.004655.
2274.
Zhou Y, Yang Z, Guo Y, Geng S, Gao S, Ye S, Hu Y, Wang Y. 2020. A New Predictor of Disease Severity in Patients with COVID-19 in Wuhan, China. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.24.20042119.
2275.
Nie S, Zhao X, Zhao K, Zhang Z, Zhang Z, Zhang Z. 2020. Metabolic disturbances and inflammatory dysfunction predict severity of coronavirus disease 2019 (COVID-19): a retrospective study. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.24.20042283.
2276.
Tan W, Lu Y, Zhang J, Wang J, Dan Y, Tan Z, He X, Qian C, Sun Q, Hu Q, Liu H, Ye S, Xiang X, Zhou Y, Zhang W, Guo Y, Wang X-H, He W, Wan X, Sun F, Wei Q, Chen C, Pan G, Xia J, Mao Q, Chen Y, Deng G. 2020. Viral Kinetics and Antibody Responses in Patients with COVID-19. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.24.20042382.
2277.
Jiang H, Li Y, Zhang H, Wang W, Men D, Yang X, Qi H, Zhou J, Tao S. 2020. Global profiling of SARS-CoV-2 specific IgG/ IgM responses of convalescents using a proteome microarray. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.20.20039495.
Zhang D, Guo R, Lei L, Liu H, Wang Y, Wang Y, Qian H, Dai T, Zhang T, Lai Y, Wang J, Liu Z, Chen T, He A, O’Dwyer M, Hu J. 2020. COVID-19 infection induces readily detectable morphological and inflammation-related phenotypic changes in peripheral blood monocytes, the severity of which correlate with patient outcome. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.24.20042655.
2280.
Miller A, Reandelar MJ, Fasciglione K, Roumenova V, Li Y, Otazu GH. 2020. Correlation between universal BCG vaccination policy and reduced morbidity and mortality for COVID-19: an epidemiological study. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.24.20042937.
BCG vaccination to reduce the impact of COVID-19 in healthcare workers (The BRACE Trial). Murdoch Children's Research Institute. https://www.mcri.edu.au/BRACE. Retrieved 31 July 2020.
2283.
Brann DH, Tsukahara T, Weinreb C, Lipovsek M, Van den Berge K, Gong B, Chance R, Macaulay IC, Chou H, Fletcher R, Das D, Street K, de Bezieux HR, Choi Y-G, Risso D, Dudoit S, Purdom E, Mill JS, Hachem RA, Matsunami H, Logan DW, Goldstein BJ, Grubb MS, Ngai J, Datta SR. 2020. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.25.009084.
2284.
Smith JC, Sausville EL, Girish V, Yuan ML, John KM, Sheltzer JM. 2020. Cigarette smoke exposure and inflammatory signaling increase the expression of the SARS-CoV-2 receptor ACE2 in the respiratory tract. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.28.013672.
2285.
Liu R, Liu X, Han H, Shereen MA, Niu Z, Li D, Liu F, Wu K, Luo Z, Zhu C. 2020. The comparative superiority of IgM-IgG antibody test to real-time reverse transcriptase PCR detection for SARS-CoV-2 infection diagnosis. Cold Spring Harbor Laboratory https://doi.org/10.1101/2020.03.28.20045765.
14 Appendix A
This appendix contains reviews produced by the Immunology Institute of the Icahn School of Medicine
14.1 Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody
Considering the relatively high identity of the receptor binding domain
(RBD) of the spike proteins from 2019-nCoV and SARS-CoV (73%), this
study aims to assess the cross-reactivity of several anti-SARS-CoV
monoclonal antibodies with 2019-nCoV. The results showed that the
SARS-CoV-specific antibody CR3022 can potently bind 2019-nCoV RBD.
14.1.3 Main Findings
The structure of the 2019-nCoV spike RBD and its conformation in complex
with the receptor angiotensin-converting enzyme (ACE2) was modeled in
silico and compared with the SARS-CoV RBD structure. The models
predicted very similar RBD-ACE2 interactions for both viruses. The
binding capacity of representative SARS-CoV-RBD specific monoclonal
antibodies (m396, CR3014, and CR3022) to recombinant 2019-nCoV RBD was
then investigated by ELISA and their binding kinetics studied using
biolayer interferometry. The analysis showed that only CR3022 was able
to bind 2019-nCoV RBD with high affinity (KD of 6.3 nM), however it did
not interfere with ACE2 binding. Antibodies m396 and CR3014, which
target the ACE2 binding site of SARS-CoV failed to bind 2019-nCoV spike
protein.
14.1.4 Limitations
The 2019-nCoV RBD largely differ from the SARS-CoV at the C-terminus
residues, which drastically impact the cross-reactivity of antibodies
described for other B beta-coronaviruses, including SARS-CoV. This study
claims that CR3022 antibody could be a potential candidate for therapy.
However, none of the antibodies assayed in this work showed
cross-reactivity with the ACE2 binding site of 2019-nCoV, essential for
the replication of this virus. Furthermore, neutralization assays with
2019-nCoV virus or pseudovirus were not performed. Although the use of
neutralizing antibodies is an interesting approach, these results
suggest that it is critical the development of novel monoclonal
antibodies able to specifically bind 2019-nCoV spike protein.
14.1.5 Credit
Review by D.L.O as part of a project by students, postdocs and faculty
at the Immunology Institute of the Icahn School of Medicine, Mount
Sinai.
14.2 Integrative Bioinformatics Analysis Provides Insight into the Molecular Mechanisms of 2019-nCoV
The authors used bioinformatics tools to identify features
of ACE2 expression in the lungs of different patent groups: healthy,
smokers, patients with chronic airway disease (i.e., COPD) or asthma.
They used gene expression data publicly available from GEO that included
lung tissues, bronchoalveolar lavage, bronchial epithelial cells, small
airway epithelial cells, or SARS-Cov infected cells.
The authors describe no significant differences in ACE2 expression in
lung tissues of Healthy, COPD, and Asthma groups (p=0.85); or in BAL of
Healthy and COPD (p=0.48); or in epithelial brushings of Healthy and
Mild/Moderate/Severe Asthma (p=0.99). ACE2 was higher in the small
airway epithelium of long-term smokers vs non-smokers (p<0.001).
Consistently, there was a trend of higher ACE2 expression in the
bronchial airway epithelial cells 24h post-acute smoking exposure
(p=0.073). Increasing ACE2 expression at 24h and 48h compared to 12h
post SARS-Cov infection (p=0.026; n=3 at each time point) was also
detected.
15 lung samples’ data from healthy participants were separated into high
and low ACE2 expression groups. “High” ACE2 expression was associated
with the following GO pathways: innate and adaptive immune responses, B
cell mediated immunity, cytokine secretion, and IL-1, IL-10, IL-6, IL-8
cytokines. The authors speculate that a high basal ACE2 expression will
increase susceptibility to SARS-CoV infection.
In 3 samples SARS-Cov infection was associated with IL-1, IL-10 and IL-6
cytokine production (GO pathways) at 24h. And later, at 48h, with T-cell
activation and T-cell cytokine production. It is unclear whether those
changes were statistically significant.
The authors describe a time course quantification of immune infiltrates
in epithelial cells infected with SARS-Cov infection. They state that in
healthy donors ACE2 expression did not correlate with the immune cell
infiltration. However, in SARS-Cov samples, at 48h they found that ACE2
correlated with neutrophils, NK-, Th17-, Th2-, Th1- cells, and DCs.
Again, while authors claim significance, the corresponding correlation
coefficients and p-values are not presented in the text or figures. In
addition, the source of the data for this analysis is not clear.
Using network analysis, proteins SRC, FN1, MAPK3, LYN, MBP, NLRC4, NLRP1
and PRKCD were found to be central (Hub proteins) in the regulating
network of cytokine secretion after coronavirus infection. Authors
conclude this indicates that these molecules were critically important
in ACE2-induced inflammatory response. Additionally, authors speculate
that the increased expression of ACE2 affected RPS3 and SRC, which were
the two hub genes involved in viral replication and inflammatory
response.
14.2.3 Limitations
The methods section is very limited and does not
describe any of the statistical analyses; and description of the
construction of the regulatory protein networks is also limited. For the
findings in Figures 2 authors claim significance, which is not supported
by p-values or coefficients. For the sample selection, would be useful
if sample sizes and some of the patients’ demographics (e.g. age) were
described.
For the analysis of high vs low ACE2 expression in healthy subjects, it
is not clear what was the cut off for ‘high’ expression and how it was
determined. Additionally, further laboratory studies are warranted to
confirm that high ACE2 gene expression would have high correlation with
the amount of ACE2 protein on cell surface. For the GO pathway analysis
significance was set at p<0.05, but not adjusted for multiple
comparisons.
There were no samples with SARS-CoV-2 infection. While SARS-Cov and
SARS-CoV-2 both use ACE2 to enter the host cells, the analysis only
included data on SARS-Cov and any conclusions about SARS-CoV2 are
limited.
Upon checking GSE accession numbers of the datasets references, two
might not be cited correctly: GSE37758 (“A spergillus niger: Control
(fructose) vs. steam-exploded sugarcane induction (SEB)”” was used in
this paper as “lung tissue” data) and GSE14700 (“Steroid Pretreatment
of Organ Donors to Prevent Postischemic Renal Allograft Failure: A
Randomized, Controlled Trial” – was used as SARS-Cov infection data).
14.2.4 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.3 Diarrhea may be underestimated: a missing link in 2019 novel coronavirus
This study examined the incidence of diarrhea in
patients infected with SARS-CoV-2 across three recently published
cohorts and found that there are statistically significant differences
by Fisher’s exact test. They report that this could be due to subjective
diagnosis criterion for diarrhea or from patients first seeking medical
care from gastroenterologist. In order to minimize nosocomial infections
arising from unsuspected patients with diarrhea and gain comprehensive
understanding of transmission routes for this viral pathogen, they
compared the transcriptional levels of ACE2 of various human tissues
from NCBI public database as well as in small intestine tissue from
CD57BL/6 mice using single cell sequencing. They show that ACE2
expression is not only increased in the human small intestine, but
demonstrate a particular increase in mice enterocytes positioned on the
surface of the intestinal lining exposed to viral pathogens. Given that
ACE2 is the viral receptor for SARS-CoV-2 and also reported to regulate
diarrhea, their data suggests the small intestine as a potential
transmission route and diarrhea as a potentially underestimated symptom
in COVID19 patients that must be carefully monitored. Interestingly,
however, they show that ACE2 expression level is not elevated in human
lung tissue.
14.3.3 Limitations
Although this study demonstrates a
statistical difference in the incidence of diarrhea across three
separate COVID19 patient cohorts, their conclusions are limited by a
small sample size. Specifically, the p-value computed by Fisher’s exact
test is based on a single patient cohort of only six cases of which 33%
are reported to have diarrhea, while the remaining two larger cohorts
with 41 and 99 cases report 3% and 2% diarrhea incidence, respectively.
Despite showing significance, they would need to acquire larger sample
sizes and cohorts to minimize random variability and draw meaningful
conclusions. Furthermore, they do not address why ACE2 expression level
is not elevated in human lung tissue despite it being a major
established route of transmission for SARS-CoV-2. It could be helpful to
validate this result by looking at ACE2 expression in mouse lung tissue.
Finally, although this study is descriptive and shows elevated ACE2
expression in small intestinal epithelial cells, it does not establish a
mechanistic link to SARS-CoV-2 infection of the host. Overall, their
claim that infected patients exhibiting diarrhea pose an increased risk
to hospital staff needs to be further substantiated.
14.3.4 Significance
This study provides a possible transmission route and a
potentially underappreciated clinical symptom for SARS-CoV-2 for better
clinical management and control of COVID19.
14.3.5 Credit
Summary generated as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.4 Specific ACE2 Expression in Cholangiocytes May Cause Liver Damage After 2019-nCoV Infection
Using both publicly available scRNA-seq dataset of liver
samples from colorectal patients and scRNA-sequencing of four liver
samples from healthy volunteers, the authors show that ACE2 is
significantly enriched in the majority of cholangiocytes (59.7 %) but
not in hepatocytes (2.6%).
14.4.3 Main Findings
Using bioinformatics approaches of
RNASeq analysis, this study reveals that ACE2 dominates in
cholangiocytes and is present at very low levels in hepatocytes.
14.4.4 Limitations
The
study does not provide mechanistic insights into how SARS-CoV-2 can
infect and replicate in cholangiocytes and the types of intrinsic
anti-viral responses induced by cholangiocytes when infected. In
addition, because the study relies on the assumption that SARS-CoV-2
infects cells only through ACE2, it cannot discount the possibility that
the virus can infect hepatocytes through mechanisms other than
ACE2-mediated entry. Furthermore, because the scRNA-seq analysis were
performed on healthy liver samples, one cannot draw any definitive
conclusions about gene expression states (including ACE2 expression in
liver cell types) in system-wide inflammatory contexts.
14.4.5 Significance
This article with other studies on liver
damage in COVID patients suggests that liver damage observed in COVID
patients is more due to inflammatory cytokines than direct infection of
the liver. Even if cholangiocytes are infectable by SARS-CoV-2 (which
was demonstrated by human liver ductal organoid study
((2166)),
published clinical data show no significant increase in bile duct injury
related indexes (i.e. alkaline phosphatase, gamma-glutamyl
transpeptidase and total bilirubin). In sum, it underscores the
importance of future studies characterizing cellular responses of
extra-pulmonary organs in the context of COVID or at least in viral lung
infections..
14.4.6 Credit
Summary generated by Chang Moon as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.5 ACE2 expression by colonic epithelial cells is associated with viral infection, immunity and energy metabolism
Colonic enterocytes primarily express ACE2. Cellular pathways associated with ACE2 expression include innate immune signaling, HLA up regulation, energy metabolism and apoptotic signaling.
14.5.3 Limitations
This is a study of colonic biopsies taken from 17 children with and without IBD and analyzed using scRNAseq to look at ACE2 expression and identify gene families correlated with ACE2 expression. The authors find ACE2 expression to be primarily in colonocytes. It is not clear why both healthy and IBD patients were combined for the analysis. Biopsies were all of children so extrapolation to adults is limited. The majority of genes found to be negatively correlated with ACE2 expression include immunoglobulin genes (IGs). IG expression will almost certainly be low in colonocytes irrespective of ACE2 expression.
14.5.4 Significance
This study performs a retrospective analysis of ACE2 expression using an RNAseq dataset from intestinal biopsies of children with and without IBD. The implications for the CoV-19 epidemic are modest, but do provide support that ACE2 expression is specific to colonocytes in the intestines. The ontological pathway analysis provides some limited insights into gene expression associated with ACE2.
14.5.5 Credit
Summary generated as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.6 The Pathogenicity of 2019 Novel Coronavirus in hACE2 Transgenic Mice
Using a transgenic human Angiotensin-converting enzyme 2
(hACE2) mouse that has previously been shown susceptible to infection by
SARS-CoV, Bao et al. create a model of pandemic 2019-nCoV strain
coronavirus. The model includes interstitial hyperplasia in lung tissue,
moderate inflammation in bronchioles and blood vessels, and histology
consistent with viral pneumonia at 3 days post infection. Wildtype did
not experience these symptoms. In addition, viral antigen and hACE2
receptor were found to co-localize the lung by immunofluorescence 3-10
days post infection only in the hACE2 infected mice.
14.6.3 Limitations
The characterization of the infection remains incomplete,
as well as lacking characterization of the immune response other than
the presence of a single antiviral antibody. Though they claim to
fulfill Koch’s postulates, they only isolate the virus and re-infect
Vero cells, rather than naive mice.
14.6.4 Significance
This paper establishes a murine model for 2019-nCoV
infection with symptoms consistent with viral pneumonia. Though not
fully characterized, this model allows in vivo analysis of viral entry
and pathology that is important for the development of vaccines and
antiviral therapeutics.
14.6.5 Credit
Review by Dan Fu Ruan, Evan Cody and Venu Pothula as part of a project by students, postdocs and faculty
at the Immunology Institute of the Icahn School of Medicine, Mount
Sinai.
14.7 Caution on Kidney Dysfunctions of 2019-nCoV Patients
Retrospective study of 59 patients assayed key function indicators
of the kidney–including urine protein, blood urea nitrogen (BUN),
plasma creatinine (Cre), and renal CT scan data.
Found that 34% of patients developed massive albuminuria on the
first day of admission, and 63% developed proteinuria during their
stay in hospital; and 19% of patients had high plasma creatinine,
especially the terminal cases.
CT analyses of 27 patients showed all patients to have abnormal
kidney damage; indicate that inflammation and edema of the renal
parenchyma very common.
14.7.3 Limitations
No analysis of immunity-dependent damage and cytokines in
blood/plasma/urine. Will be worth correlating disease progression
with cytokine production, immune activity and kidney function.
Extrapolating to earlier SARS-CoV studies provides the only
rationale for viral-damage in kidney and resultant pathologic immune
response (understandable for this clinical study).
14.7.4 Significance
Multiple lines of evidence along this study’s finding point to the
idea that renal impairment/injury is a key risk factor in 2019-nCoV
patients similar to what has been reported for SARS-CoV(2170); this
may be one of the major causes of virally-induced damage and
contribute to multiorgan failure.
ACE2 expression in kidney proximal tubule epithelia and bladder
epithelia (2171) support
these clinical findings.
Study argues for closely monitoring kidney function, and applying
potential interventions including continuous renal replacement
therapies (CRRT) for protecting kidney functions as early as
possible, particularly for those with rising plasma creatinine.
14.7.5 Credit
Review by Samarth Hegde as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.8 Profiling the immune vulnerability landscape of the 2019 Novel Coronavirus
This study harnesses bioinformatic profiling to predict the potential of
COV2 viral proteins to be presented on MHC I and II and to form linear
B-cell epitopes. These estimates suggest a T-cell antigenic profile
distinct from SARS-CoV or MERS-CoV, identify focused regions of the
virus with a high density of predicted epitopes, and provide preliminary
evidence for adaptive immune pressure in the genetic evolution of the
virus.
14.8.3 Limitations
While the study performs a comprehensive analysis of potential epitopes
within the virus genome, the analysis relies solely on bioinformatic
prediction to examine MHC binding affinity and B-cell epitope potential
and does not capture the immunogenicity or recognition of
these epitopes. Future experimental validation in data from patients
infected with SARS-CoV-2 will be important to validate and refine these
findings. Thus some of the potential conclusions stated, including viral
evolution toward lower immunogenicity or a dominant role for CD4+
T-cells rather than CD8+ T-cells in viral clearance, require further
valiadtion.
14.8.4 Significance
These findings may help direct peptide vaccine design toward relevant
epitopes and provide intriguing evidence of viral evolution in response
to immune pressure.
14.8.5 Credit
Summary generated as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.9 Single-cell Analysis of ACE2 Expression in Human Kidneys and Bladders Reveals a Potential Route of 2019-nCoV Infection
To investigate the possible cause of kidney damage in 2019-nCoV
patients, authors used published kidney and bladder cell atlas data
(GSE131685, GSE108097; 3 healthy donors each) as well as an
unpublished kidney single-cell RNA-Seq data (in-house from 2
transplant donors) to evaluate ACE2 gene expressions in all cell
types of healthy kidneys and bladders.
They find enriched expression of ACE2 transcript in all subtypes of
proximal tubule cells of kidney, with 5%-15% of both straight and
convoluted proximal tubule cells expressing ACE2.
They also find detectable levels of ACE2 in bladder epithelial
cells, noting expression from around 1.5% of cells in the outer
layer umbrella cells of the bladder epithelium and decreasing in the
basal cells.
Importantly endothelial or immune cells in kidney/bladder do not
express ACE2.
14.9.3 Limitations
This study primarily characterizes ACE2 expression (amongst other
genes) from a small healthy-donor dataset, and will benefit from
supporting data in (expired) patient samples to show functional
viral damage. ACE2 transcript does not necessarily translate to
viral permissiveness in kidney/bladder epithelia or cytokine
release.
This study focuses on only healthy tissue; it will be useful to
analyze kidney/bladder epithelial ACE2 expression under inflammatory
conditions or in patients with underlying kidney conditions.
Given what is known about protease TMPRSS2 expression during
SARS-CoV-2 infection, ACE2+TMPRSS2+ double-positive cell
identification would be useful in these datasets.
14.9.4 Significance
ACE2 protein is spatially restricted to brush border of proximal
tubules and in bladder umbrella cells (67), such cells in direct
contact with viral particles are likely to be highly sensitive to
viral-induced damage.
SARS-CoV and MERS-CoV have been shown to be detected in urine of
patients and associate with higher mortality (2170, 2173), thus worth
understanding kidney damage and resultant immune response in
SARS-CoV-2 as well.
This study argues for a potential mode of viral infectivity and
resultant inflammatory responses in these tissue in addition to
reported infectivity in the lung and digestive system, which is
supported by clinical data showing acute and early kidney
complications in 2019-nCoV patients (2169).
Clinically, thus very important to track urinary CoVID-19 shedding
as well as study acute kidney injury-related co-morbidities.
14.9.5 Credit
Review by Samarth Hegde as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.10 Neutrophil-to-Lymphocyte Ratio Predicts Severe Illness Patients with 2019 Novel Coronavirus in the Early Stage
This study aimed to find prognostic biomarkers of COVID-19
pneumonia severity. Sixty-one (61) patients with COVID-19 treated in
January at a hospital in Beijing, China were included. On average,
patients were seen within 5 days from illness onset. Samples were
collected on admission; and then patients were monitored for the
development of severe illness with a median follow-up of 10 days].
Patients were grouped as “mild” (N=44) or “moderate/severe” (N=17)
according to symptoms on admission and compared for different
clinical/laboratory features. “Moderate/severe” patients were
significantly older (median of 56 years old, compared to 41 years old).
Whereas comorbidies rates were largely similar between the groups,
except for hypertension, which was more frequent in the severe group (p=
0.056). ‘Severe’ patients had higher counts of neutrophils, and serum
glucose levels; but lower lymphocyte counts, sodium and serum chlorine
levels. The ratio of neutrophils to lymphocytes (NLR) was also higher
for the ‘severe’ group. ‘Severe’ patients had a higher rate of bacterial
infections (and antibiotic treatment) and received more intensive
respiratory support and treatment.
26 clinical/laboratory variables were used to select NLR and age as the
best predictors of the severe disease. Predictive cutoffs for a severe
illness as NLR ≥ 3.13 or age ≥ 50 years.
14.10.3 Limitations
Identification of early biomarkers is important for
making clinical decisions, but large sample size and validation cohorts
are necessary to confirm findings. It is worth noting that patients
classified as “mild” showed pneumonia by imaging and fever, and in
accordance with current classifications this would be consistent with
“moderate” cases. Hence it would be more appropriate to refer to the
groups as “moderate” vs “severe/critical”. Furthermore, there are
several limitations that could impact the interpretation of the results:
e.g. classification of patients was based on symptoms presented on
admission and not based on disease progression, small sample size,
especially the number of ‘severe’ cases (with no deaths among these
patients). Given the small sample size, the proposed NLR and age cut
offs might not hold for a slightly different set of patients. For
example, in a study of >400 patients, ‘non-severe’ and ‘severe’ NLR
were 3.2 and 5.5, respectively (2175).
14.10.4 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.11 Characteristics of lymphocyte subsets and cytokines in peripheral blood of 123 hospitalized patients with 2019 novel coronavirus pneumonia (NCP)
The authors analyzed lymphocyte subsets and cytokines of 102 patients
with mild disease and 21 with severe disease. CD8+T cells and CD4+T
cells were significantly reduced in both cohort. particularly in severe
patients. The cytokines IL6 and IL10 were significantly elevated in
severe patients as compared to mild. No significant differences were
observed in frequency of B cells and NK cells.
The authors argue that the measurement of T cell frequencies and
cytokine levels of IL6 and IL10 can be used to predict progression of
disease from Mild to severe Cov-2 infection.
14.11.3 Limitations
The study demonstrates in a limited cohort
similar associations to several other reported studies. The authors
didn’t compare the changes in lymphocyte and cytokine with healthy
individual (Covid-19 Negative) rather used an internal standard value.
The recently preprint in LANCET shows The degree of lymphopenia and a
pro-inflammatory cytokine storm is higher in severe COVID-19 patients
than in mild cases, and is associated with the disease severity (2177).
14.11.4 Significance
This translational data identifies key cytokines and lymphopenia
associated with disease severity although mechanism and key cellular
players are still unknown. Higher level IL-6 production in severe
patient suggests potential role of Tocilizumab (anti-IL6R) biologic
although clinical trial will be necessary.
14.11.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.12 Epidemiological and Clinical Characteristics of 17 Hospitalized Patients with 2019 Novel Coronavirus Infections Outside Wuhan, China
These authors looked at 17 hospitalized patients with COVID-19
confirmed by RT-PCR in Dazhou, Sichuan. Patients were admitted between
January 22 and February 10 and the final data were collected on February
11. Of the 17 patients, 12 remained hospitalized while 5 were discharged
after meeting national standards. The authors observed no differences
based on the sex of the patients but found that the discharged patients
were younger in age (p = 0.026) and had higher lymphocyte counts (p =
0.005) and monocyte counts (p = 0.019) upon admission.
14.12.3 Limitations
This study is limited in the sample size of the study and the last
data collection point was only one day after some of the patients were
admitted.
14.12.4 Significance
These findings have been somewhat supported by subsequent studies
that show that older age and an immunocompromised state are more likely
to result in a more severe clinical course with COVID-19. However, other
studies have been published that report on larger numbers of cases.
14.12.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.13 ACE2 Expression in Kidney and Testis May Cause Kidney and Testis Damage After 2019-nCoV Infection
Study used online datasets (scRNAseq GSE131685, scRNAseq GSE107585,
Human Protein Atlas, GTEx portal, CCLE) to analyze ACE2 expression
in different human organs.
Study re-analyzed three clinical datasets (n=6, n=99, and n=41) to
show 3~10% of 2019-nCoV patients present with abnormal renal
function.
results indicate ACE2 highly expressed in renal tubular cells,
Leydig cells and seminiferous ductal cells of testis.
14.13.3 Limitations
Very preliminary transcript/protein dataset analysis in healthy
cohorts; does not necessarily translate to actual viral tropism and
permissiveness.
Clinically, would be important to determine with larger longitudinal
dataset if SARS-CoV-2 infection changes sperm quality or testicular
inflammation.
Similarly, would be important to determine if simultaneous HBV or
syphilis infection and orchitis impacts SARS-CoV-2 severity.
Examination and follow-up of renal function and viral orchitis/sperm
quality of CoVID-19 patients not done in this preliminary study.
14.13.4 Significance
Kidney ACE2 result supports other concurrent sequencing studies
(2171) and clinical reports
of abnormal renal function or even kidney damage in patients
infected with 2019-nCoV
(2169).
High ACE2 expression in testis suggests potential tropism of the
virus to testicular tissues and indicates potential risks for male
fertility. Viral orchitis reported for SARS-CoV previously [1],
but no clear evidence so far of infertility in SARS, MERS or
CoVID-19 patients.
14.13.5 Credit
Review by Samarth Hegde as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.14 Aberrant pathogenic GM-CSF+ T cells and inflammatory CD14+CD16+ monocytes in severe pulmonary syndrome patients of a new coronavirus
The authors of this study sought to characterize the immune
mechanism causing severe pulmonary disease and mortality in 2019-nCoV
(COVID-19) patients. Peripheral blood was collected from hospitalized
ICU (n=12) and non-ICU (n=21) patients with confirmed 2019-nCoV and from
healthy controls (n=10) in The First Affiliated Hospital of University
of Science and Technology China (Hefei, Anhui). Immune analysis was
conducted by flow cytometry. 2019-nCoV patients had decreased
lymphocyte, monocyte, and CD4 T cell counts compared to healthy
controls. ICU patients had fewer lymphocytes than non-ICU patients. CD4
T cells of 2019-nCoV patients expressed higher levels of activation
markers (OX40, CD69, CD38, CD44) and exhaustion markers (PD-1 and Tim3)
than those of healthy controls. CD4 cells of ICU patients expressed
significantly higher levels of OX40, PD-1, and Tim3 than those of
non-ICU patients. 2019-nCoV patients had higher percentages of CD4 T
cells co-expressing GM-CSF and IL-6 compared to healthy controls, while
ICU patients had a markedly higher percentage of GM-CSF+ IFN-γ+ CD4 T
cells than non-ICU patients. The CD4 T cells of nCoV patients and
healthy controls showed no differences in TNF-α secretion.
The CD8 T cells of 2019-nCoV patients also showed higher expression of
activation markers CD69, CD38, and CD44, as well as exhaustion markers
PD-1 and Tim3, compared to healthy controls. CD8 T cells of ICU patients
expressed higher levels of GM-CSF than those of non-ICU patients and
healthy controls. No IL-6 or TNF-α was found in the CD8 T cells of any
group. There were no differences in numbers of NK cells or B cells in
2019-nCoV patients and healthy controls, nor was there any GM-CSF or
IL-6 secretion from these cells in either group.
Percentages of CD14+ CD16+ GM-CSF+ and CD14+ CD16+ IL-6+ inflammatory
monocytes were significantly increased in nCoV patients compared to
healthy controls; in particular, patients in the ICU had greater
percentages of CD14+ CD16+ IL-6+ monocytes than non-ICU patients. The
authors suggest that in 2019-nCoV patients, pathogenic Th1 cells produce
GM-CSF, recruiting CD14+ CD16+ inflammatory monocytes that secrete high
levels of IL-6. These may enter pulmonary circulation and damage lung
tissue while initiating the cytokine storm that causes mortality in
severe cases. This is consistent with the cytokine storm seen in similar
coronaviruses, as IL-6, IFN-γ, and GM-CSF are key inflammatory mediators
seen in patients with SARS-CoV-1 and MERS-CoV.
14.14.3 Limitations
Though the results of this study open
questions for further investigation, this is an early study on a small
cohort of patients, and as such there are a number of limitations. The
study included only 12 ICU patients and 21 non-ICU patients, and ideally
would be repeated with a much larger patient cohort. Though the authors
make claims about differences in lymphocyte and monocyte counts between
patients and healthy controls, they did not report baseline laboratory
findings for the control group. Additionally, severity of disease was
classified based on whether or not patients were in the ICU. It would be
interesting to contextualize the authors’ immunological findings with
more specific metrics of disease severity or time course. Noting
mortality, time from disease onset, pre-existing conditions, or severity
of lung pathology in post-mortem tissue samples would paint a fuller
picture of how to assess risk level and the relationship between
severity of disease and immunopathology. Another limitation is the
selection of cytokines and immune markers for analysis, as the selection
criteria were based on the cell subsets and cytokine storm typically
seen in SARS-CoV-1 and MERS-CoV patients. Unbiased cytokine screens and
immune profiling may reveal novel therapeutic targets that were not
included in this study.
14.14.4 Significance
This study identifies potential therapeutic targets
that could prevent acute respiratory disease syndrome (ARDS) and
mortality in patients most severely affected by COVID-19. The authors
propose testing monoclonal antibodies against IL6-R or GM-CSF to block
recruitment of inflammatory monocytes and the subsequent cytokine storm
in these patients.
14.14.5 Credit
Review by Gabrielle Lubitz as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.15 Clinical Characteristics of 2019 Novel Infected Coronavirus Pneumonia:A Systemic Review and Meta-analysis
The authors performed a meta analysis of literature on clinical,
laboratory and radiologic characteristics of patients presenting with
pneumonia related to SARSCoV2 infection, published up to Feb 6 2020.
They found that symptoms that were mostly consistent among studies were
sore throat, headache, diarrhea and rhinorrhea. Fever, cough, malaise
and muscle pain were highly variable across studies. Leukopenia (mostly
lymphocytopenia) and increased white blood cells were highly variable
across studies. They identified three most common patterns seen on CT
scan, but there was high variability across studies. Consistently across
the studies examined, the authors found that about 75% of patients need
supplemental oxygen therapy, about 23% mechanical ventilation and about
5% extracorporeal membrane oxygenation (ECMO). The authors calculated a
staggering pooled mortality incidence of 78% for these patients.
14.15.3 Limitations
The authors mention that the total number of studies included in this
meta analysis is nine, however they also mentioned that only three
studies reported individual patient data. It is overall unclear how many
patients in total were included in their analysis. This is mostly
relevant as they reported an incredibly high mortality (78%) and mention
an absolute number of deaths of 26 cases overall. It is not clear from
their report how the mortality rate was calculated.
The data is based on reports from China and mostly from the Wuhan area,
which somewhat limits the overall generalizability and applicability of
these results.
14.15.4 Significance
This meta analysis offers some important data for clinicians to refer to
when dealing with patients with COVID-19 and specifically with
pneumonia. It is very helpful to set expectations about the course of
the disease.
14.15.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.16 Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients
Liu et al. enrolled a cohort of 40 patients from Wuhan
including 27 mild cases and 13 severe cases of COVID-19. They performed
a 16-day kinetic analysis of peripheral blood from time of disease
onset. Patients in the severe group were older (medium age of 59.7,
compared to 48.7 in mild group) and more likely to have hypertension as
a co-morbidity. Lymphopenia was observed in 44.4% of the mild patients
and 84.6% of the severe patients. Lymphopenia was due to low T cell
count, specially CD8 T cells. Severe patients showed higher neutrophil
counts and an increase of cytokines in the serum (IL2, IL6, IL10 and
IFNγ). The authors measured several other clinical laboratory parameters
were also higher in severe cases compared to mild, but concluded that
neutrophil to CD8 T cell ratio (N8R) as the best prognostic factor to
identify the severe cases compared to other receiver operating
characteristic (ROC).
14.16.3 Limitations
This was a small cohort (N=40), and two of the patients
initially included in the severe group (N=13) passed away and were
excluded from the analysis due to lack of longitudinal data. However, it
would be most important to be able to identify patients with severe
disease with higher odds of dying. It seems that the different time
points analyzed relate to hospital admission, which the authors describe
as disease onset. The time between first symptoms and first data points
is not described. It would have been important to analyze how the
different measured parameters change according to health condition, and
not just time (but that would require a larger cohort). The predictive
value of N8R compared to the more commonly used NLR needs to be assessed
in other independent and larger cohorts. Lastly, it is important to note
that pneumonia was detected in patients included in the “mild” group,
but according to the Chinese Clinical Guidance for COVID-19 Pneumonia
Diagnosis and Treatment (7th edition) this group should be considered
“moderate”.
14.16.4 Significance
Lymphopenia and cytokine storm have been described to be
detrimental in many other infections including SARS-CoV1 and MERS-CoV.
However, it was necessary to confirm that this dramatic immune response
was also observed in the SARS-CoV2 infected patients. These results and
further validation of the N8R ratio as a predictor of disease severity
will contribute for the management of COVID19 patients and potential
development of therapies.
14.16.5 Credit
Review by Pauline Hamon as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.17 Clinical and immunologic features in severe and moderate forms of Coronavirus Disease 2019
This study retrospectively evaluated clinical, laboratory,
hematological, biochemical and immunologic data from 21 subjects
admitted to the hospital in Wuhan, China (late December/January) with
confirmed SARS-CoV-2 infection. The aim of the study was to compare
‘severe’ (n=11, ~64 years old) and ‘moderate’ (n=10, ~51 years old)
COVID-19 cases. Disease severity was defined by patients’ blood oxygen
level and respiratory output. They were classified as ‘severe’ if SpO2
93% or respiratory rates 30 per min.
In terms of the clinical laboratory measures, ‘severe’ patients had
higher CRP and ferritin, alanine and aspartate aminotransferases, and
lactate dehydrogenase but lower albumin concentrations.
The authors then compared plasma cytokine levels (ELISA) and immune cell
populations (PBMCs, Flow Cytometry). ‘Severe’ cases had higher levels of
IL-2R, IL-10, TNFa, and IL-6 (marginally significant). For the immune
cell counts, ‘severe’ group had higher neutrophils, HLA-DR+ CD8 T cells
and total B cells; and lower total lymphocytes, CD4 and CD8 T cells
(except for HLA-DR+), CD45RA Tregs, and IFNy-expressing CD4 T cells. No
significant differences were observed for IL-8, counts of NK cells,
CD45+RO Tregs, IFNy-expressing CD8 T and NK cells.
14.17.3 Limitations
Several potential limitations should be noted: 1) Blood samples were
collected 2 days post hospital admission and no data on viral loads were
available; 2) Most patients were administered medications (e.g.
corticosteroids), which could have affected lymphocyte counts.
Medications are briefly mentioned in the text of the manuscript; authors
should include medications as part of Table 1. 3) ‘Severe’ cases were
significantly older and 4/11 ‘severe’ patients died within 20 days.
Authors should consider a sensitivity analysis of biomarkers with the
adjustment for patients’ age.
14.17.4 Significance
Although the sample size was small, this paper presented a broad range
of clinical, biochemical, and immunologic data on patients with
COVID-19. One of the main findings is that SARS-CoV-2 may affect T
lymphocytes, primarily CD4+ T cells, resulting in decreased IFNy
production. Potentially, diminished T lymphocytes and elevated cytokines
can serve as biomarkers of severity of COVID-19.
14.17.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.18 SARS-CoV-2 and SARS-CoV Spike-RBD Structure and Receptor Binding Comparison and Potential Implications on Neutralizing Antibody and Vaccine Development
This study compared the structure of SARS-CoV and SARS-CoV-2 Spike (S)
protein receptor binding domain (RBD) and interactions with ACE2 using
computational modeling, and interrogated cross-reactivity and
cross-neutralization of SARS-CoV-2 by antibodies against SARS-CoV. While
SARS-CoV and SARS-CoV-2 have over 70 % sequence homology and share the
same human receptor ACE2, the receptor binding motif (RBM) is only 50%
homologous.
Computational prediction of the SARS-CoV-2 and ACE2 interactions based
on the previous crystal structure data of SARS-CoV, and measurement of
binding affinities against human ACE2 using recombinant SARS-CoV and
SARS-CoV-2 S1 peptides, demonstrated similar binding of the two S1
peptides to ACE2, explaining the similar transmissibility of SARS-CoV
and SARS-CoV-2 and consistent with previous data (Wall et al Cell
2020).
The neutralization activity of SARS-CoV-specific rabbit polyclonal
antibodies were about two-order of magnitude less efficient to
neutralize SARS-CoV-2 than SARS-CoV, and four potently neutralizing
monoclonal antibodies against SARS-CoV had poor binding and neutralizing
activity against SARS-CoV-2. In contrast, 3 poor SARS-CoV-binding
monoclonal antibodies show some efficiency to bind and neutralize
SARS-CoV-2. The results suggest that that antibodies to more conserved
regions outside the RBM motif might possess better cross-protective
neutralizing activities between two strains.
14.18.3 Limitations
It would have been helpful to show the epitopes recognized by the
monoclonal antibodies tested on both SARS-CoV, SARS-CoV-2 to be able to
make predictions for induction of broadly neutralizing antibodies. The
data on monoclonal antibody competition with ACE2 for binding to
SARS-CoV RBD should have also included binding on SARS-CoV2, especially
for the three monoclonal antibodies that showed neutralization activity
for SARS-CoV2. Because of the less homology in RBM sequences between
viruses, it still may be possible that these antibodies would recognize
the ACE2 RBD in SARS-CoV-2.
14.18.4 Significance
It is noteworthy that immunization to mice and rabbit with SARS-CoV S1
or RBD protein could induce monoclonal antibodies to cross-bind and
cross-neutralize SARS-CoV-2 even if they are not ACE2-blocking. If these
types of antibodies could be found in human survivors or in the
asymptomatic populations as well, it might suggest that exposure to
previous Coronavirus strains could have induced cross-neutralizing
antibodies and resulted in the protection from severe symptoms in some
cases of SARS-CoV2.
14.18.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.19 Protection of Rhesus Macaque from SARS-Coronavirus challenge by recombinant adenovirus vaccine
Rhesus macaques were immunized intramuscularly twice (week 0 and
week 4) with SV8000 carrying the information to express a S1-orf8
fusion protein and the N protein from the BJ01 strain of SARS-CoV-1.
By week 8, immunized animals had signs of immunological protection
(IgG and neutralization titers) against SARS-CoV-1 and were
protected against challenge with the PUMC-1 strain, with fewer
detectable symptoms of respiratory distress, lower viral load,
shorter periods of viral persistence, and less pathology in the
lungs compared to non-immunized animals.
14.19.3 Limitations
The authors should write clearer descriptions of the methods used in
this article. They do not describe how the IgG titers or
neutralization titers were determined. There are some issues with
the presentation of data, for example, in Figure 1a, y-axis should
not be Vmax; forming cells and 1d would benefit from showing error
bars. Furthermore, although I inferred that the animals were
challenged at week 8, the authors did not explicitly detail when the
animals were challenged. The authors should explain the design of
their vaccine, including the choice of antigens and vector. The
authors also do not include a description of the ethical use of
animals in their study.
14.19.4 Significance
The authors describe a vaccine for SARS-CoV-1 with no discussion of
possible implications for the current SARS-CoV-2 pandemic. Could a
similar vaccine be designed to protect against SARS-CoV-2 and would
the concerns regarding emerging viral mutations that the authors
describe as a limitation for SARS-CoV-1 also be true in the context
of SARS-CoV-2?
14.19.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.20 Reduction and Functional Exhaustion of T cells in Patients with Coronavirus Disease 2019 (COVID-19)
Based on a retrospective study of 522 COVID patients and 40
healthy controls from two hospitals in Wuhan, China, authors show both
age-dependent and clinical severity-dependent decrease in T cell numbers
with elderly patients and patients who are in ICU-care showing the most
dramatic decrease in T cell counts. Cytokine profiling of COVID patients
reveal that TNF-α, IL-6 and IL-10 are increased in infected patients
with patients in the ICU showing the highest levels. Interestingly,
these three cytokine levels were inversely correlated with T cell counts
and such inverse relationship was preserved throughout the disease
progression. Surface staining of exhaustion markers (PD-1 and Tim-3) and
flow cytometry of stained peripheral blood of 14 patients and 3 healthy
volunteers demonstrate that T cells of COVID patients have increased
expression of PD-1 with patients in ICU having the highest number of
CD8+PD-1+ cells than their counterparts in non-ICU groups.
14.20.3 Limitations
Compared to the number of patients, number of control (n=
40) is small and is not controlled for age. Additional data linking
inflammatory cytokines and the quality of the adaptive response
including humoral and antigen specific T cell response is much needed. T
cell exhaustion study relies on marker-dependent labeling of T cell
functionality of a very limited sample size (n=17)—a
functional/mechanistic study of these T cells from PBMCs would have
bolstered their claims.
14.20.4 Significance
Limited but contains interesting
implications. It is already known in literature that in the context of
acute respiratory viral infections CD8 T cells exhibit exhaustion-like
phenotypes which further underscores the importance of mechanistic
studies that can elucidate how COVID infection leads to lymphopenia and
T cell exhaustion-like phenotype.
However, as authors have noted, the data does point to an interesting
question: How these inflammatory cytokines (TNF-α, IL-6 and IL-10)
correlate with or affect effective viral immunity and what types of
cells produce these cytokines? Answering that question will help us
refine our targets for immune-modulatory therapies especially in
patients suffering from cytokine storms.
14.20.5 Credit
This review by Chang Moon was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.21 Clinical Characteristics of 25 death cases infected with COVID-19 pneumonia: a retrospective review of medical records in a single medical center, Wuhan, China
Most common chronic conditions among 25 patients that died from
COVID-19 related respiratory failure were hypertension (64%) and
diabetes (40%). Disease progression was marked by progressive organ
failure, starting first with lung dysfunction, then heart (e.g.
increased cTnI and pro-BNP), followed by kidney (e.g. increased BUN,
Cr), and liver (e.g. ALT, AST). 72% of patients had neutrophilia and 88%
also had lymphopenia. General markers of inflammation were also
increased (e.g. PCT, D-Dimer, CRP, LDH, and SAA).
14.21.3 Limitations
The limitations of this study include small sample size and
lack of measurements for some tests for several patients. This study
would also have been stronger with comparison of the same measurements
to patients suffering from less severe disease to further validate and
correlate proposed biomarkers with disease severity.
14.21.4 Significance
This study identifies chronic conditions (i.e. hypertension
and diabetes) that strongly correlates with disease severity. In
addition to general markers of inflammation, the authors also identify
concomitant neutrophilia and lymphopenia among their cohort of patients.
This is a potentially interesting immunological finding because we would
typically expect increased lymphocytes during a viral infection.
Neutrophilia may also be contributing to cytokine storm. In addition,
PCT was elevated in 90.5% of patients, suggesting a role for sepsis or
secondary bacterial infection in COVID-19 related respiratory failure.
14.21.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.22 SARS-CoV-2 infection does not significantly cause acute renal injury: an analysis of 116 hospitalized patients with COVID-19 in a single hospital, Wuhan, China
Clinical data from 116 hospitalized CoVID-19 patients analyzed over
4 weeks for correlation with renal injury. Comorbidities included
chronic renal failure (CRF) in 5 patients (4.3%).
10.8% of patients with no prior kidney disease showed elevations in
blood urea or creatinine, and 7.2% of patients with no prior kidney
disease showed albuminuria.
Patients with pre-existing CRF underwent continuous renal
replacement therapy (CRRT) alongside CoVID-19 treatment. Renal
functions remained stable in these patients.
All 5 patients with CRF survived CoVID-19 therapy without
progression to ARDS or worsening of CRF.
14.22.3 Limitations
Renal injury biomarkers in patients with incipient kidney
abnormalities not tabulated separately, making overall data hard to
interpret. It will be critical to separately examine kidney function
(BUN, urine creatinine and eGFR) in patients that developed any
kidney abnormalities (7.2~10.8% of cohort).
No information on type of CoVID-19 therapy used across cohort; will
be useful to correlate how treatment modality influences kidney
function (and other parameters).
Invokes previous clinical-correlation studies that indicate low
instances of kidney damage(2188, 2189), but those studies did not track
longitudinal urine samples for acute renal injury markers and viral
shedding.
CRRT in patients with CRF is standard therapy irrespective of
CoVID-19 status; it will be important to compare clinical parameters
of these patients (n=5) with virus-naïve CRF patients (none in this
study) to make any meaningful conclusions.
14.22.4 Significance
This study argues that renal impairment is uncommon in CoVID-19 and
not associated with high mortaility, in stark contrast with a
concurrent study (2169).
If supported by further studies, this argues kidney impairment is
secondary to cytokine storm/inflammation-induced organ failure, and
not due to direct viral replication.
Will be important to comprehensively characterize large-datasets of
CoVID-19 patients to conclude if kidney function actively disrupted
due to viral infection.
14.22.5 Credit
Review by Samarth Hegde as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.23 Potential T-cell and B-Cell Epitopes of 2019-nCoV
The authors use 2 neural network algorithms, NetMHCpan4 and
MARIA, to identify regions within the COVID-19 genome that are
presentable by HLA. They identify 405 viral epitopes that are
presentable on MHC-I and MHC-II and validate using known epitopes from
SARS-CoV. To determine whether immune surveillance drives viral
mutations to evade MHC presentation, the authors analyzed 68 viral
genomes from 4 continents. They identified 93 point mutations that
occurred preferentially in regions predicted to be presented by MHC-I
(p=0.02) suggesting viral evolution to evade CD8 T-cell mediated
killing. 2 nonsense mutations were also identified that resulted in loss
of presentation of an associated antigen (FGDSVEEVL) predicted to be
good antigen for presentation across multiple HLA alleles.
To identify potential sites of neutralizing antibody binding, the
authors used homology modeling to the SARS-CoV’s spike protein (S
protein) to determine the putative structure of the CoV2 spike protein.
They used Discotope2 to identify antibody binding sites on the protein
surface in both the down and up conformations of the S protein. The
authors validate this approach by first identifying antibody binding
site in SARS-CoV S protein. In both the down and up conformation of the
CoV2 S protein, the authors identified a potential antibody binding site
on the S protein receptor binding domain (RBD) of the ACE2 receptor
(residues 440-460, 494-506). While RBDs in both SARS-CoV and CoV2 spike
proteins may be important for antibody binding, the authors note that
SARS-CoV has larger attack surfaces than CoV2. These results were later
validated on published crystal structures of the CoV2 S protein RBD and
human ACE2. Furthermore, analysis of 68 viral genomes did not identify
any mutations in this potential antibody binding site in CoV2.
Finally, the authors compile a list of potential peptide vaccine
candidates across the viral genome that can be presented by multiple HLA
alleles. Several of the peptides showed homology to SARS-CoV T-cell and
B-cell epitopes.
14.23.3 Limitations
While the authors used computational methods of validation,
primarily through multiple comparisons to published SARS-CoV structures
and epitopes, future work should include experimental validation of
putative T-cell and B-cell epitopes.
14.23.4 Significance
The authors identified potential T-cell and B-cell epitopes
that may be good candidates for peptide based vaccines against CoV2.
They also made interesting observations in comparing SARS-CoV and CoV2
potential antibody binding sites, noting that SARS-CoV had larger attack
surfaces for potential neutralizing antibody binding. One of the
highlights of this paper was the authors’ mutation analysis of 68 viral
genomes from 4 continents. This analysis not only validated their
computational method for identifying T-cell epitopes, but showed that
immune surveillance likely drives viral mutation in MHC-I binding
peptides. The smaller attack surface may point to potential mechanisms
of immune evasion by CoV2. However, absence of mutations in the RBD of
CoV2 and the small number of mutations in peptides presentable to T
cells suggests that vaccines against multiple epitopes could still
elicit robust immunity against CoV2.
14.23.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.24 Structure, Function, and Antigenicity of the SARSCoV-2 Spike Glycoprotein
The authors highlight a human angiotensin-converting enzyme 2
(hACE2), as a potential receptor used by the current Severe Acute
respiratory syndrome coronavirus-2 (SARS-CoV-2) as a host factor that
allows the virus target human cells. This virus-host interaction
facilitates the infection of human cells with a high affinity comparable
with SARS-CoV. The authors propose this mechanism as a probable
explanation of the efficient transmission of SARS-CoV-2 between humans.
Besides, Walls and colleagues described SARS-CoV-2 S glycoprotein S by
Cryo-EM along with neutralizing polyclonal response against SAR-CoV-2 S
from mice immunized with SAR-CoV and blocking SAR-CoV-2 S-mediated entry
into VeroE6 infected cells.**
14.24.3 Limitations
The SARS-CoV-2 depends on the cell factors ACE2 and TMPRSS2, this
last, according to a recent manuscript by Markus Hoffman et al., Cell,
2020. The authors used green monkey (VeroE6) and hamster (BHK) cell
lines in the experiments to drive its conclusions to humans; however, it
is well known the caucasian colon adenocarcinoma human cell line
(CaCo-2), highly express the hACE2 receptor as the TMPRSS2 protease as
well. In humans, ACE2 protein is highly expressed in the
gastrointestinal tract, which again, makes the CaCo-2 cell line suitable
for the following SARS-CoV-2 studies.
14.24.4 Significance
The results propose a functional receptor used by SARS-CoV-2 to
infect humans worldwide and defining two distinct conformations of spike
(S) glycoprotein by cryogenic electron microscopy (Cryo-EM). This study
might help establish a precedent for initial drug design and treatment
of the current global human coronavirus epidemic.
14.24.5 Credit
Review by postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.25 Breadth of concomitant immune responses underpinning viral clearance and patient recovery in a non-severe case of COVID-19
The authors characterized the immune response in peripheral blood of a
47-year old COVID-19 patient.
SARS-CoV2 was detected in nasopharyngeal swab, sputum and faeces
samples, but not in urine, rectal swab, whole blood or throat swab. 7
days after symptom onset, the nasopharyngeal swab test turned negative,
at day 10 the radiography infiltrates were cleared and at day 13 the
patient became asymptomatic.
Immunofluorescence staining shows from day 7 the presence of
COVID-19-binding IgG and IgM antibodies in plasma, that increase
until day 20.
Flow cytometry on whole blood reveals a plasmablast peak at day 8, a
gradual increase in T follicular helper cells, stable HLA-DR+ NK
frequencies and decreased monocyte frequencies compared to healthy
counterparts. The expression of CD38 and HLA-DR peaked on T cells at D9
and was associated with higher production of cytotoxic mediators by
CD8+ T cells.
IL-6 and IL-8 were undetectable in plasma.
The authors further highlight the presence of the IFITM3
SNP-rs12252-C/C variant in this patient, which is associated with
higher susceptibility to influenza virus.
14.25.3 Limitations
These results need to be confirmed in additional patients.
COVID-19 patients have increased infiltration of macrophages in their
lungs (2193). Monitoring monocyte proportions in blood earlier in the
disease might help to evaluate their eventual migration to the lungs.
The stable concentration of HLA-DR+ NK cells in blood from day 7 is
not sufficient to rule out NK cell activation upon SARS-CoV2 infection.
In response to influenza A virus, NK cells express higher levels of
activation markers CD69 and CD38, proliferate better and display higher
cytotoxicity (2194). Assessing these parameters in COVID-19 patients is
required to better understand NK cell role in clearing this infection.
Neutralization potential of the COVID-19-binding IgG and IgM antibodies
should be assessed in future studies.
This patient was able to clear the virus, while presenting a SNP
associated with severe outcome following influenza infection. The
association between this SNP and outcome upon SARS-CoV2 infection should
be further investigated.
14.25.4 Significance
This study is among the first to describe the appearance of
COVID-19-binding IgG and IgM antibodies upon infection. The emergence of
new serological assays might contribute to monitor more precisely the
seroconversion kinetics of COVID-19 patients (2195). Further association
studies between IFITM3 SNP-rs12252-C/C variant and clinical data might
help to refine the COVID-19 outcome prediction tools.
14.25.5 Credit
Review by Bérengère Salomé as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.26 The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing
The authors performed single-cell RNA sequencing (scRNAseq) on
bronchoalveolar lavage fluid (BAL) from 6 COVID-19 patients (n=3 mild
cases, n=3 severe cases). Data was compared to previously generated
scRNAseq data from healthy donor lung tissue (n=8).
Clustering analysis of the 6 patients revealed distinct immune cell
organization between mild and severe disease. Specifically, they found
that transcriptional clusters annotated as tissue resident alveolar
macrophages were strongly reduced while monocytes-derived FCN1+SPP1+
inflammatory macrophages dominated the BAL of patients with severe
COVID19 diseases. They show that inflammatory macrophages upregulated
interferon-signaling genes, monocytes recruiting chemokines including
CCL2, CCL3, CCL4 as well as IL-6, TNF, IL-8 and profibrotic cytokine
TGF-β, while alveolar macrophages expressed lipid metabolism genes, such
as PPARG.
The lymphoid compartment was overall enriched in lungs from patients.
Clonally expanded CD8 T cells were enriched in mild cases suggesting
that CD8 T cells contribute to viral clearance as in Flu infection,
whereas proliferating T cells were enriched in severe cases.
SARS-CoV-2 viral transcripts were detected in severe patients, but
considered here as ambient contaminations.
14.26.3 Limitations
These results are based on samples from 6 patients and should therefore
be confirmed in the future in additional patients. Longitudinal
monitoring of BAL during disease progression or resolution would have
been most useful.
The mechanisms underlying the skewing of the macrophage compartment in
patients towards inflammatory macrophages should be investigated in
future studies.
Deeper characterization of the lymphoid subsets is required. The
composition of the “proliferating” cluster and how these cells differ
from conventional T cell clusters should be assessed. NK and CD8 T cell
transcriptomic profile, in particular the expression of cytotoxic
mediator and immune checkpoint transcripts, should be compared between
healthy and diseased lesions.
14.26.4 Significance
COVID-19 induces a robust inflammatory cytokine storm in patients that
contributes to severe lung tissue damage and ARDS (2196). Accumulation of
monocyte-derived inflammatory macrophages at the expense of Alveolar
macrophages known to play an anti-inflammatory role following
respiratory viral infection, in part through the PPARγ pathway (2197, 2198) are
likely contributing to lung tissue injuries. These data suggest that
reduction of monocyte accumulation in the lung tissues could help
modulate COVID-19-induced inflammation. Further analysis of lymphoid
subsets is required to understand the contribution of adaptive immunity
to disease outcome.
14.26.5 Credit
Review by Bérengère Salomé and Assaf Magen as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.27 Can routine laboratory tests discriminate 2019 novel coronavirus infected pneumonia from other community-acquired pneumonia?
In an attempt to use standard laboratory testing for the discrimination
between “Novel Coronavirus Infected Pneumonia” (NCIP) and a usual
community acquired pneumonia (CAP), the authors compared laboratory
testing results of 84 NCIP patients with those of a historical group of
316 CAP patients from 2018 naturally COVID-19 negative. The authors
describe significantly lower white blood- as well as red blood- and
platelet counts in NCIP patients. When analyzing differential blood
counts, lower absolute counts were measured in all subsets of NCIP
patients. With regard to clinical chemistry parameters, they found
increased AST and bilirubin in NCIP patients as compared to CAP
patients.
14.27.3 Limitations
The authors claim to describe a simple method to rapidly assess a
pre-test probability for NCIP. However, the study has substantial
weakpoints. The deviation in clinical laboratory values in NCIP patients
described here can usually be observed in severely ill patients. The
authors do not comment on how severely ill the patients tested here were
in comparison to the historical control. Thus, the conclusion that the
tests discriminate between CAP and NCIP lacks justification.
14.27.4 Significance
The article strives to compare initial laboratory testing results in
patients with COVID-19 pneumonia as compared to patients with a usual
community acquired pneumonia. The implications of this study for the
current clinical situation seem restricted due to a lack in clinical
information and the use of a control group that might not be
appropriate.
14.27.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.28 Correlation Analysis Between Disease Severity and Inflammation-related Parameters in Patients with COVID-19 Pneumonia
This study is a cross-sectional analysis of 100 patients with
COVID-19 pneumonia, divided into mild (n = 34), severe (n = 34), and
critical (n = 32) disease status based on clinical definitions.
The criteria used to define disease severity are as follows:
Severe – any of the following: respiratory distress or respiratory
rate ≥ 30 respirations/minute; oxygen saturation ≤ 93% at rest;
oxygen partial pressure (PaO2)/oxygen concentration (FiO2) in
arterial blood ≤ 300mmHg, progression of disease on imaging
to >50% lung involvement in the short term.
Critical – any of the following: respiratory failure that requires
mechanical ventilation; shock; other organ failure that requires
treatment in the ICU.
Patients with pneumonia who test positive for COVID-19 who do not
have the symptoms delineated above are considered mild.
Peripheral blood inflammatory markers were correlated to disease status.
Disease severity was significantly associated with levels of IL-2R,
IL-6, IL-8, IL-10, TNF-α, CRP, ferroprotein, and procalcitonin. Total
WBC count, lymphocyte count, neutrophil count, and eosinophil count were
also significantly correlated with disease status. Since this is a
retrospective, cross-sectional study of clinical laboratory values,
these data may be extrapolated for clinical decision making, but without
studies of underlying cellular causes of these changes this study does
not contribute to a deeper understanding of SARS-CoV-2 interactions with
the immune system.
It is also notable that the mean age of patients in the mild group was
significantly different from the mean ages of patients designated as
severe or critical (p < 0.001). The mean patient age was not
significantly different between the severe and critical groups. However,
IL-6, IL-8, procalcitonin (Table 2), CRP, ferroprotein (Figure 3A, 3B),
WBC count, and neutrophil count (Figure 4A, 4B) were all significantly
elevated in the critical group compared to severe. These data suggest
underlying differences in COVID-19 progression that is unrelated to age.
14.28.3 Significance
Given the inflammatory profile outlined in this study,
patients who have mild or severe COVID-19 pneumonia, who also have any
elevations in the inflammatory biomarkers listed above, should be
closely monitored for potential progression to critical status.
14.28.4 Credit
This review by JJF was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.29 An Effective CTL Peptide Vaccine for Ebola Zaire Based on Survivors’ CD8+ Targeting of a Particular Nucleocapsid Protein Epitope with Potential Implications for COVID-19 Vaccine Design
Vaccination of mice with a single dose of a 9-amino-acid peptide NP44-52
located in a conserved region of ebolavirus (EBOV) nucleocapsid protein
(NP) confers CD8+ T-cell-mediated immunity against mouse adapted EBOV
(maEBOV). Bioinformatic analyses predict multiple conserved CD8+ T cell
epitopes in the SARS-CoV-2 NP, suggesting that a similar approach may be
feasible for vaccine design against SARS-CoV-2.
The authors focus on a site within a 20-peptide region of EBOV NP which
was commonly targeted by CD8+ T cells in a group of EBOV survivors
carrying the HLA-A*30:01:01 allele. To justify the testing of specific
vaccine epitopes in a mouse challenge setting, the authors cite known
examples of human pathogen-derived peptide antigens that are also
recognized by C57BL/6 mice, as well as existing data surrounding known
mouse immunogenicity of peptides related to this EBOV NP region. Testing
3 distinct 9mer peptides over an 11 amino-acid window and comparing to
vaccination with the 11mer with a T-cell reactivity readout demonstrated
that optimizing peptide length and position for immunogenicity may be
crucial, likely due to suboptimal peptide processing and MHC-class-I
loading.
Vaccines for maEBOV challenge studies were constructed by packaging
NP44-52 in d,l poly(lactic-co-glycolic) acid microspheres. CpG was also
packaged within the microspheres, while Monophosphoryl Lipid A (a TLR4
ligand) was added to the injectate solution. A second peptide consisting
of a predicted MHC-II epitope from the EBOV VG19 protein was added using
a separate population of microspheres, and the formulation was injected
by intraperitoneal administration. The vaccine was protective against a
range of maEBOV doses up to at least 10,000 PFU. Survival was
anticorrelated with levels of IL6, MCP-1 (CCL2), IL9, and GM-CSF, which
recapitulated trends seen in human EBOV infection.
While HLA-A*30:01:01 is only present in a minority of humans, the
authors state that MHC binding algorithms predict NP44-52 to be a strong
binder of a set of more common HLA-A*02 alleles. The authors predict
that a peptide vaccine based on the proposed formulation could elicit
responses in up to 50% of people in Sudan or 30% of people in North
America.
SARS-CoV-2 NP, meanwhile, has conserved regions which may provide
peptide-vaccine candidates. Scanning the SARS-CoV-2 NP sequence for
HLA-binding 9mers identified 53 peptides with predicted binding affinity
< 500nM, including peptides that are predicted to bind to HLA-class-I
alleles of 97% of humans, 7 of which have previously been tested
in-vitro.
The results support previously appreciated correlations between certain
cytokines and disease severity, specifically IL6 which relates to
multiple trial therapies. Prediction of HLA-class-I binding of
SARS-CoV-2 NP peptides suggests the plausibility of a peptide vaccine
targeting conserved regions of SARS-CoV-2 NP although further validation
in previously infected patient samples will be essential.
14.29.3 Credit
Review by Andrew M. Leader as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.30 Epitope-based peptide vaccines predicted against novel coronavirus disease caused by SARS-CoV-2
This study employs a series of bioinformatic
pipelines to identify T and B cell epitopes on spike (S) protein of
SARS-CoV-2 and assess their properties for vaccine potential. To
identify B cell epitopes, they assessed structural accessibility,
hydrophilicity, and beta-turn and flexibility which are all factors that
promote their targeting by antibodies. To identify T cell epitopes, they
filtered for peptides with high antigenicity score and capacity to bind
3 or more MHC alleles. Using the protein digest server, they also
demonstrated that their identified T and B cell epitopes are stable,
having multiple non-digesting enzymes per epitope. Epitopes were also
determined to be non-allergenic and non-toxin as assessed by Allergen FP
1.0 and ToxinPred, respectively. For T cell epitopes, they assessed the
strength of epitope-HLA interaction via PepSite. Overall, they predict
four B cell and eleven T cell epitopes (two MHC I and nine MHC II
binding) to pass stringent computational thresholds as candidates for
vaccine development. Furthermore, they performed sequence alignment
between all identified SARS-CoV-2 S protein mutations and predicted
epitopes, and showed that the epitopes are conserved across 134 isolates
from 38 locations worldwide. However, they report that these conserved
epitopes may soon become obsolete given the known mutation rate of
related SARS-CoV is estimated to be 4x10-4/site/year, underscoring the
urgency of anti-viral vaccine development.
14.30.3 Limitations
While spike (S) protein may have a
critical role in viral entry into host cells and their epitope
prediction criterion were comprehensive, this study did not examine
other candidate SARS-CoV-2 proteins. This point is particularly
important given that a single epitope may not be sufficient to induce
robust immune memory, and recent approaches involve multi-epitope
vaccine design. Furthermore, their study only included a direct
implementation of various published methods, but did not validate
individual bioinformatic tools with controls to demonstrate robustness.
Finally, it is critical that these predicted epitopes are experimentally
validated before any conclusions can be drawn about their potential as
vaccine candidates or their clinical efficacy.
14.30.4 Significance
This study provides a computational framework to
rapidly identify epitopes that may serve as potential vaccine candidates
for treating SARS-CoV-2.
14.30.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.31 The definition and risks of Cytokine Release Syndrome-Like in 11 COVID-19-Infected Pneumonia critically ill patients: Disease Characteristics and Retrospective Analysis
This study describes the occurrence of a cytokine release syndrome-like
(CRSL) toxicity in ICU patients with COVID-19 pneumonia. The median time
from first symptom to acute respiratory distress syndrome (ARDS) was 10
days. All patients had decreased CD3, CD4 and CD8 cells, and a
significant increase of serum IL-6. Furthermore, 91% had decreased NK
cells. The changes in IL-6 levels preceded those in CD4 and CD8 cell
counts. All of these parameters correlated with the area of pulmonary
inflammation in CT scan images. Mechanical ventilation increased the
numbers of CD4 and CD8 cells, while decreasing the levels of IL-6, and
improving the immunological parameters.
14.31.3 Limitations
The number of patients included in this retrospective single center
study is small (n=11), and the follow-up period very short (25 days).
Eight of the eleven patients were described as having CRSL, and were
treated by intubation (7) or ECMO (2). Nine patients were still in the
intensive care unit at the time of publication of this article, so their
disease outcome is unknown.
14.31.4 Significance
The authors define a cytokine release syndrome-like toxicity in patients
with COVID-19 with clinical radiological and immunological criteria: 1)
decrease of circulating CD4, CD8 and NK cells; 2) substantial increase
of IL-6 in peripheral blood; 3) continuous fever; 4) organ and tissue
damage. This event seems to occur very often in critically ill patients
with COVID-19 pneumonia. Interestingly, the increase of IL-6 in the
peripheral blood preceded other laboratory alterations, thus, IL-6 might
be an early biomarker for the severity of COVID-19 pneumonia. The
manuscript will require considerable editing for organization and
clarity.
14.31.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.32 Clinical characteristics of 36 non-survivors with COVID-19 in Wuhan, China
This is a simple study reporting clinical
characteristics of patients who did not survive COVID-19. All patients
(mean age=69.22 years) had acute respiratory distress syndrome (ARDS)
and their median time from onset to ARDS was 11 days. The median time
from onset to death was 17 days. Most patients were older male (70%
male) with co-morbidities and only 11 % were smokers. 75% patients
showed bilateral pneumonia. Many patients had chronic diseases,
including hypertension (58.33%). cardiovascular disease (22.22%) and
diabetes (19.44%). Typical clinical feature measured in these patients
includes lymphopenia and elevated markers of inflammation.
14.32.3 Limitations
As noted by the authors, the conclusions of this study are
very limited because this is single-centered study focusing on a small
cohort of patients who did not survive. Many clinical parameters
observed by the authors (such* as increase levels of serum CRP, PCT,
IL-6) have also been described in other COVID19 patients who survived
the infection
14.32.4 Significance
This study is essentially descriptive and may be useful for
clinical teams monitoring COVID19 patients.
14.32.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.33 Risk Factors Related to Hepatic Injury in Patients with Corona Virus Disease 2019
Based on a retrospective study of 85 hospitalized COVID
patients in a Beijing hospital, authors showed that patients with
elevated ALT levels (n = 33) were characterized by significantly higher
levels of lactic acid and CRP as well as lymphopenia and hypoalbuminemia
compared to their counterparts with normal ALT levels. Proportion of
severe and critical patients in the ALT elevation group was
significantly higher than that of normal ALT group. Multivariate
logistic regression performed on clinical factors related to ALT
elevation showed that CRP \(\geq\) 20mg/L and low lymphocyte count
(<1.1*10^9 cells/L) were independently related to ALT elevation—a
finding that led the authors to suggest cytokine storm as a major
mechanism of liver damage.
14.33.3 Limitations
The article’s most attractive claim that liver damage seen in COVID
patients is caused by cytokine storm (rather than direct infection of
the liver) hinges solely on their multivariate regression analysis.
Without further mechanistic studies a) demonstrating how high levels of
inflammatory cytokines can induce liver damage and b) contrasting types
of liver damage incurred by direct infection of the liver vs.
system-wide elevation of inflammatory cytokines, their claim remains
thin. It is also worth noting that six of their elevated ALT group
(n=33) had a history of liver disease (i.e. HBV infection, alcoholic
liver disease, fatty liver) which can confound their effort to pin down
the cause of hepatic injury to COVID.
14.33.4 Significance
Limited. This article confirms a rich body
of literature describing liver damage and lymphopenia in COVID patients.
14.33.5 Credit
Review by Chang Moon as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.34 Detectable serum SARS-CoV-2 viral load (RNAaemia) is closely associated with drastically elevated interleukin 6 (IL-6) level in critically ill COVID-19 patients
48 adult patients diagnosed with Covid19 according to Chinese guidelines
for Covid19 diagnosis and treatment version 6 were included in this
study. Patients were further sub-divided into three groups based on
clinical symptoms and disease severity: (1) mild, positive Covid19 qPCR
with no or mild clinical symptoms (fever; respiratory; radiological
abnormalities); (2) severe, at least one of the following: shortness of
breath/respiratory rate >30/min, oxygen saturation SaO2<93%,
Horowitz index paO2/FiO2 < 300 mmHg (indicating moderate pulmonary
damage); and (3) critically ill, at least one additional complicating
factor: respiratory failure with need for mechanical ventilation;
systemic shock; multi-organ failure and transfer to ICU. Serum samples
and throat-swaps were collected from all 48 patients enrolled.
SARS-CoV-2 RNA was assessed by qPCR with positive results being defined
as Ct values < 40, and serum interleukin-6 (IL-6) was quantified
using a commercially available detection kit. Briefly, patient
characteristics in this study confirm previous reports suggesting that
higher age and comorbidities are significant risk factors of clinical
severity. Of note, 5 out of 48 of patients (10.41%), all in the
critically ill category, were found to have detectable serum SARS-CoV-2
RNA levels, so-called RNAaemia. Moreover, serum IL-6 levels in these
patients were found to be substantially higher and this correlated with
the presence of detectable SARS-CoV-2 RNA levels. The authors
hypothesize that viral RNA might be released from acutely damages
tissues in moribund patients during the course of Covid19 and that
RNaemia along with IL-6 could potentially be used as a prognostic
marker.
14.34.3 Limitations
While this group’s report generally confirms some of the major findings
of a more extensive study, published in early February 2020, (2196),
there are limitations that should be taken into account. First, the
number of patients enrolled is relatively small; second, interpretation
of these data would benefit from inclusion of information about study
specifics as well as providing relevant data on the clinical course of
these patients other than the fact that some were admitted to ICU (i.e.
demographics on how many patients needed respiratory support, dialysis,
APACHE Ii/III or other standard ICU scores as robust prognostic markers
for mortality etc). It also remains unclear at which time point the
serum samples were taken, i.e. whether at admission, when the diagnosis
was made or during the course of the hospital stay (and potentially
after onset of therapy, which could have affected both IL-6 and RNA
levels). The methods section lacks important information on the qPCR
protocol employed, including primers and cycling conditions used. From a
technical point of view, Ct values >35 seem somewhat non-specific
(although Ct <40 was defined as the CDC cutoff as well) indicating
that serum RNA levels are probably very low, therefore stressing the
need for highly specific primers and high qPCR efficiency. In addition,
the statistical tests used (t-tests, according to the methods section)
do not seem appropriate as the organ-specific data such as BUN and
troponin T values seem to be not normally distributed across groups (n=
5 RNAaemia+ vs. n= 43 RNAaemia-). Given the range of standard deviations
and the differences in patient sample size, it is difficult to believe
that these data are statistically significantly different.
14.34.4 Significance
This study is very rudimentary and lacks a lot of relevant clinical
details. However, it corroborates some previously published observations
regarding RNAemia and IL-6 by another group. Generally, regarding future
studies, it would be important to address the question of IL-6 and other
inflammatory cytokine dynamics in relation to Covid19 disease kinetics
(high levels of IL-6, IL-8 and plasma leukotriene were shown to have
prognostic value at the onset of ARDS ; serum IL-2 and IL-15 have been
associated with mortality; reviewed by Chen W & Ware L, Clin Transl Med.
2015 (2207)).
14.34.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.35 Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study
Based on a retrospective study of 162 COVID patients from a
local hospital in Wuhan, China, the authors show an inverse correlation
between lymphocyte % (LYM%) of patients and their disease severity. The
authors have also tracked LYM% of 70 cases (15 deaths; 15 severe; 40
moderate) throughout the disease progression with fatal cases showing no
recovery of lymphocytes ( <5%) even after 17-19 days post-onset. The
temporal data of LYM % in COVID patients was used to construct a
Time-Lymphocyte% model which is used to categorize and predict patients’
disease severity and progression. The model was validated using 92
hospitalized cases and kappa statistic test was used to assess agreement
between predicted disease severity and the assigned clinical severity (k
= 0.49).
14.35.3 Limitations
Time-Lymphocyte % Model (TLM) that authors have proposed as
a predictive model for clinical severity is very simple in its
construction and derives from correlative data of 162 patients. In order
for the model to be of use, it needs validation using a far more robust
data set and possibly a mechanistic study on how COVID leads to
lymphopenia in the first place. In addition, it should be noted that no
statistical test assessing significance of LYM % values between disease
severities was performed.
14.35.4 Significance
This article is of limited significance as
it simply reports similar descriptions of COVID patients made in
previous literature that severe cases are characterized by lymphopenia.
14.35.5 Credit
Review by Chang Moon as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.36 The potential role of IL-6 in monitoring severe case of coronavirus disease 2019
Study on blood biomarkers on 80 COVID19 patients (69 severe
and 11 non-severe). Patients with severe symptoms at admission
(baseline) showed obvious lymphocytopenia and significantly increased
interleukin-6 (IL-6) and CRP, which was positively correlated with
symptoms severity. IL-6 at baseline positively correlates with CRP, LDH,
ferritin and D-Dimer abundance in blood.
Longitudinal analysis of 30 patients (before and after treatment) showed
significant reduction of IL-6 in remission cases.
14.36.3 Limitations
Limited sample size at baseline, especially for the
non-severe leads to question on representativeness. The longitudinal
study method is not described in detail and suffers from
non-standardized treatment. Limited panel of pro-inflammatory cytokine
was analyzed. Patients with severe disease show a wide range of altered
blood composition and biomarkers of inflammation, as well as differences
in disease course (53.6% were cured, about 10% developed acute
respiratory distress syndrome). The authors comment on associations
between IL-6 levels and outcomes, but these were not statistically
significant (maybe due to the number of patients, non-standardized
treatments, etc.) and data is not shown. Prognostic biomarkers could
have been better explored. Study lacks multivariate analysis.
14.36.4 Significance
IL-6 could be used as a pharmacodynamic
marker of disease severity. Cytokine Release Syndrome (CRS) is a
well-known side effect for CAR-T cancer therapy and there are several
effective drugs to manage CRS. Drugs used to manage CRS could be tested
to treat the most severe cases of COVID19.
14.36.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.37 Clinical and Laboratory Profiles of 75 Hospitalized Patients with Novel Coronavirus Disease 2019 in Hefei, China
The authors of this study provide a comprehensive analysis of clinical
laboratory assessments in 75 patients (median age 47 year old)
hospitalized for Corona virus infection in China measuring differential
blood counts including T-cell subsets (CD4, CD8), coagulation function,
basic blood chemistry, of infection-related biomarkers including CRP,
Procalcitonin (PCT) (Precursor of calcitonin that increases during
bacterial infection or tissue injury), IL-6 and erythrocyte
sedimentation rate as well as clinical parameters. Among the most common
hematological changes they found increased neutrophils, reduced CD4 and
CD8 lymphocytes, increased LDH, CRP and PCT
When looking at patients with elevated IL-6, the authors describe
significantly reduced CD4 and CD8 lymphocyte counts and elevated CRP and
PCT levels were significantly increased in infected patients suggesting
that increased IL-6 may correlate well with disease severity in COVID-19
infections
14.37.3 Limitations
The authors performed an early assessment of clinical standard
parameters in patients infected with COVID-19. Overall, the number of
cases (75) is rather low and the snapshot approach does not inform about
dynamics and thus potential relevance in the assessment of treatment
options in this group of patients.
14.37.4 Significance
The article summarizes provides a good summary of some of the common
changes in immune cells inflammatory cytokines in patients with a
COVID-19 infection and. Understanding how these changes can help predict
severity of disease and guide therapy including IL-6 cytokine receptor
blockade using Tocilizumab or Sarilumab will be important to explore.
14.37.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.38 Exuberant elevation of IP-10, MCP-3 and IL-1ra during SARS-CoV-2 infection is associated with disease severity and fatal outcome
Plasma cytokine analysis (48 cytokines) was
performed on COVID-19 patient plasma samples, who were sub-stratified as
severe (N=34), moderate (N=19), and compared to healthy controls (N=8).
Patients were monitored for up to 24 days after illness onset: viral
load (qRT-PCR), cytokine (multiplex on subset of patients), lab tests,
and epidemiological/clinical characteristics of patients were reported.
14.38.3 Main Findings
Many elevated cytokines with COVID-19 onset compared to healthy
controls (IFNy, IL-1Ra, IL-2Ra, IL-6, IL-10, IL-18, HGF, MCP-3, MIG,
M-CSF, G-CSF, MIG-1a, and IP-10).
IP-10, IL-1Ra, and MCP-3 (esp. together) were associated with
disease severity and fatal outcome.
IP-10 was correlated to patient viral load (r=0.3006, p=0.0075).
IP-10, IL-1Ra, and MCP-3 were correlated to loss of lung function
(PaO2/FaO2 (arterial/atmospheric O2) and Murray Score (lung
injury) with MCP-3 being the most correlated (r=0.4104 p<0.0001
and r=0.5107 p<0.0001 respectively).
Viral load (Lower Ct Value from qRT-PCR) was associated with
upregulated IP-10 only (not IL-1Ra or MCP-3) and was mildly
correlated with decreased lung function: PaO2/FaO2
(arterial/atmospheric O2) and Murray Score (lung injury).
Lymphopenia (decreased CD4 and CD8 T cells) and increased neutrophil
correlated w/ severe patients.
Complications were associated with COVID severity (ARDS, hepatic
insufficiency, renal insufficiency).
14.38.4 Limitations
Collection time of clinical data and lab results
not reported directly (likely 4 days (2,6) after illness onset), making
it very difficult to determine if cytokines were predictive of patient
outcome or reflective of patient compensatory immune response (likely
the latter). Small N for cytokine analysis (N=2 fatal and N=5
severe/critical, and N=7 moderate or discharged). Viral treatment
strategy not clearly outlined.
14.38.5 Expanded Results
NOTE: Moderate COVID-19 was classified by fever, respiratory
manifestations, and radiological findings consistent with pneumonia
while severe patients had one or more of the following: 1) respiratory
distraction, resting O2 saturation, or 3) arterial PaO2/FiO2 < 300
mmHg.
Significant elevation of cytokines observed in COVID patients
compared to healthy controls: IFNy, IL-1Ra, IL-2Ra, IL-6, IL-10,
IL-18, HGF, MCP-3, MIG, M-CSF, G-CSF, MIG-1a, and IP-10.
Severity was correlated with increase in measured IP-10, MCP-3,
and IL-Ra as measure by area under the curve analysis during
sample timecourse (2-24 days after illness onset).
IL-1Ra incr. significant 0-7 days after onset, MCP-3 signif.
upregulated throughout observation timecourse, and IP-10 increased
and upregulated throughout (trending downwards over time).
The three cytokines together (IP-10, IL-1Ra, and MCP-3 AUC) served
as the best predictors of disease deterioration and fatal outcome.
No significant differences between moderate/severe observed between
groups in IL-2Ra, IL-6, IL-10, IL-18, CTACK, G-CSF, HGF, M-CSF,
MIP-1a, MIG, and IFNy at any timepoints.
Viral load (Lower Ct Value from qRT-PCR) was associated with
upregulated IP-10 only (not IL-1Ra or MCP-3) and was highly
correlated with decreased lung function: PaO2/FaO2
(arterial/atmospheric O2) and Murray Score (lung injury).
Antibodies against these cytokines (esp. anti-IP-10) may serve as
a potential treatment for amelioration of COVID-19 (and associated
ARDS).
Lab results:
Decreased lymphocytes (%) in all patients – lymphopenia corr. w/
severe patients
Decreased CD4 and CD8 T cells – no monocyte or
eosino/basophil % measured
Increased neutrophils (%)
Increased BUN (mmol/L) – other kidney markers, liver markers, and
LDH were not significantly different between groups and were not
compared to healthy controls.
Clinical features (between moderate vs. severe patient groups):
Complications were associated with severity (ARDS, hepatic
insufficiency, renal insufficiency).
Coexisting conditions between groups were not significantly
different (chronic heart/lung/renal/liver disease, diabetes, or
cancer) and patient time courses (onset to admission and onset to
viral tx) also not significantly different – 4 days (2, 6) on
average for admission and 4 (3,7) for antiviral.
Increased corticosteroids and mechanical/ invasive mechanical
ventilation in severe patients.
Increased median age in severe group (Median (Range = 63.5 (42-74)
vs. 51 (22-78)) and patients > 60 yrs had higher ratio of
severe patients as compared patients 16-59 yrs.
Higher incidence of fever is severe patients (91.2 vs. 68.4%),
myalgia (57.7 vs. 48.1%), and chill (17.6% vs. 0%).
No differences in cough, headache, nausea/vomiting, or diarrhea.
14.38.6 Significance
Outline of pathological time course (implicating innate
immunity esp.) and identification key cytokines associated with disease
severity and prognosis (+ comorbidities). Anti-IP-10 as a possible
therapeutic intervention (ex: Eldelumab).
14.38.7 Credit
Review by Natalie Vaninov as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.39 Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019
This study examined antibody responses in
the blood of COVID-19 patients during the early SARS CoV2 outbreak in
China. Total 535 plasma samples were collected from 173 patients (51.4%
female) and were tested for seroconversion rate using ELISA. Authors
also compared the sensitivity of RNA and antibody tests over the course
of the disease . The key findings are:
Among 173 patients, the seroconversion rates for total antibody
(Ab), IgM and IgG were 93.1% (161/173), 82.7% (143/173) and 64.7%
(112/173), respectively.
The seroconversion sequentially appeared for Ab, IgM and then IgG,
with a median time of 11, 12 and 14 days, respectively. Overall, the
seroconversion of Ab was significantly quicker than that of IgM (p =
0.012) and IgG (p < 0.001). Comparisons of seroconversion rates
between critical and non-critical patients did not reveal any
significant differences.
RNA tests had higher sensitivity in early phase and within 7 days of
disease onset than antibody assays (66.7% Vs 38.3% respectively).
The sensitivity of the Ab assays was higher 8 days after disease
onset, reached 90% at day 13 and 100% at later time points (15-39
days). In contrast, RNA was only detectable in 45.5% of samples at
days 15-39.
In patients with undetectable RNA in nasal samples collected during
day 1-3, day 4-7, day 8-14 and day 15-39 since disease onset, 28.6%
(2/7), 53.6% (15/28), 98.2% (56/57) and 100% (30/30) had detectable
total Ab titers respectively Combining RNA and antibody tests
significantly raised the sensitivity for detecting COVID-19 patients
in different stages of the disease (p < 0.001).
There was a strong positive correlation between clinical severity
and antibody titer 2-weeks after illness onset.
Dynamic profiling of viral RNA and antibodies in representative
COVID-19 patients (n=9) since onset of disease revealed that
antibodies may not be sufficient to clear the virus. It should be
noted that increases in of antibody titers were not always
accompanied by RNA clearance.
14.39.3 Limitations
Because different types of ELISA assays were used for
determining antibody concentrations at different time points after
disease onset, sequential seroconversion of total Ab, IgM and IgG may
not represent actual temporal differences but rather differences in the
affinities of the assays used. Also, due to the lack of blood samples
collected from patients in the later stage of illness, how long the
antibodies could last remain unknown. For investigative dynamics of
antibodies, more samples were required.
14.39.4 Significance
Total and IgG antibody titers could be used to understand
the epidemiology of SARS CoV-2 infection and to assist in determining
the level of humoral immune response in patients.
The findings provide strong clinical evidence for routine serological
and RNA testing in the diagnosis and clinical management of COVID-19
patients. The understanding of antibody responses and their half-life
during and after SARS CoV2 infection is important and warrants further
investigations.
14.39.5 Credit
This review was undertaken by Zafar Mahmood and edited by K Alexandropoulos as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.40 Restoration of leukomonocyte counts is associated with viral clearance in COVID-19 hospitalized patients
The authors collected data on 25 COVID-19 patients (n=11 men, n=14
women) using standard laboratory tests and flow cytometry. All patients
were treated with antibiotics. Twenty-four of the 25 patients were also
treated with anti-viral Umefinovir and 14 of the patients were treated
with corticosteroids. 14 patients became negative for the virus after
8-14 days of treatment. The same treatment course was extended to 15-23
days for patients who were still positive for the virus at day 14.
The authors found a negative association between age and resolution of
infection. Patients with hypertension, diabetes, malignancy or chronic
liver disease were all unable to clear the virus at day 14, though not
statistically significant.
Elevated procalcitonin and a trend for increased IL-6 were also found in
peripheral blood prior to the treatment.
A trend for lower NK cell, T cell and B cell counts in patients was also
reported. B cell, CD4 and CD8 T cell counts were only increased upon
treatment in patients who cleared the virus. NK cell frequencies
remained unchanged after treatment in all the patients.
14.40.3 Limitations
73% of the patients who remained positive for SARS-CoV2 after the 1st
treatment, and 43% of all patients who cleared the virus were treated
with corticosteroids. Corticosteroids have strong effects on the immune
compartment in blood (2214). The authors should have accounted for
corticosteroid treatment when considering changes in T, NK and B cell
frequencies.
Assessing if IL-6 concentrations were back to baseline levels following
treatment would have provided insights into the COVID-19 cytokine storm
biology. Patients with higher baseline levels of IL-6 have been reported
to have lower CD8 and CD4 T cell frequencies (2210). Correlating IL-6 with
cell counts before and after treatment would thus have also been of
interest. The report of the laboratory measures in table 2 is incomplete
and should include the frequencies of patients with increased/decreased
levels for each parameter.
Correction is needed for the 1st paragraph of the discussion as data
does not support NK cell restoration upon treatment in patients who
cleared the virus. NK cells remain unchanged after the 1st treatment
course and only seem to increase in 2 out of 6 donors after the 2nd
treatment course in those patients.
14.40.4 Significance
Previous reports suggest an association between disease severity and
elevated IL-6 or pro-calcitonin concentrations in COVID-19
patients (2206, 2215). IL-6 receptor blockade is also being administered to
patients enrolled in clinical trials (NCT04317092). This report thus
contributes to highlight elevated concentrations of these analytes in
COVID-19 patients. Mechanisms underlying the association between viral
clearance and restoration of the T cell and B cell frequencies suggests
viral-driven immune dysregulation, which needs to be investigated in
further studies.
14.40.5 Credit
Review by Bérengère Salomé as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.41 Clinical findings in critically ill patients infected with SARS-CoV-2 in Guangdong Province, China: a multi-center, retrospective, observational study
This work analyses laboratory and clinical data from 45 patients treated
in the in ICU in a single province in China. Overall, 44% of the
patients were intubated within 3 days of ICU admission with only 1
death.
Lymphopenia was noted in 91% of patient with an inverse correlation with
LDH.
Lymphocyte levels are negatively correlated with Sequential Organ
Failure Assessment (SOFA) score (clinical score, the higher the more
critical state), LDH levels are positively correlated to SOFA score.
Overall, older patients (>60yo), with high SOFA score, high LDH
levels and low lymphocytes levels at ICU admission are at higher risk of
intubation.
Of note, convalescent plasma was administered to 6 patients but due to
limited sample size no conclusion can be made.
14.41.3 Limitations
While the study offers important insights into disease course and
clinical lab correlates of outcome, the cohort is relatively small and
is likely skewed towards a less-severe population compared to other ICU
reports given the outcomes observed. Analysis of laboratory values and
predictors of outcomes in larger cohorts will be important to make
triage and treatment decisions. As with many retrospective analyses,
pre-infection data is limited and thus it is not possible to understand
whether lymphopenia was secondary to underlying comorbidities or
infection.
Well-designed studies are necessary to evaluate the effect of
convalescent plasma administration.
14.41.4 Significance
This clinical data enables the identification of at-risk
patients and gives guidance for research for treatment options. Indeed,
further work is needed to better understand the causes of the
lymphopenia and its correlation with outcome.
14.41.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.42 Immune Multi-epitope vaccine design using an immunoinformatics approach for 2019 novel coronavirus in China (SARS-CoV-2)
Using in silico bioinformatic tools, this study identified putative
antigenic B-cell epitopes and HLA restricted T-cell epitopes from the
spike, envelope and membrane proteins of SARS-CoV-2, based on the genome
sequence available on the NCBI database. T cell epitopes were selected
based on predicted affinity for the more common HLA-I alleles in the
Chinese population. Subsequently, the authors designed vaccine peptides
by bridging selected B-cell epitopes and adjacent T-cell epitopes.
Vaccine peptides containing only T-cell epitopes were also generated.
From 61 predicted B-cell epitopes, only 19 were exposed on the surface
of the virion and had a high antigenicity score. A total of 499 T-cell
epitopes were predicted. Based on the 19 B-cell epitopes and their 121
adjacent T-cell epitopes, 17 candidate vaccine peptides were designed.
Additionally, another 102 vaccine peptides containing T-cell epitopes
only were generated. Based on the epitope counts and HLA score, 13 of
those were selected. Thus, a total of 30 peptide vaccine candidates were
designed.
14.42.3 Limitations
While this study provides candidates for the development of vaccines
against SARS-CoV-2, in vitro and in vivo trials are required to validate
the immunogenicity of the selected B and T cell epitopes. This could be
done using serum and cells from CoV-2-exposed individuals, and in
preclinical studies. The implication of this study for the current
epidemic are thus limited. Nevertheless, further research on this field
is greatly needed.
14.42.4 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.43 Clinical Features of Patients Infected with the 2019 Novel Coronavirus (COVID-19) in Shanghai, China
This single-center cohort study analyzes the clinical and
laboratory features of 198 patients with confirmed COVID-19 infection in
Shanghai, China and correlated these parameters with clinical disease
severity, including subsequent intensive care unit (ICU) admission. 19
cases (9.5%) required ICU admission after developing respiratory failure
or organ dysfunction. Age, male sex, underlying cardiovascular disease,
and high symptom severity (high fever, dyspnea) were all significantly
correlated with ICU admission. Additionally, ICU admission was more
common in patients who presented with lymphopenia and elevated
neutrophil counts, among other laboratory abnormalities. Flow cytometric
analysis revealed that patients admitted to the ICU had significantly
reduced circulating CD3+ T cell, CD4+ T cell, CD8+ T cell, and CD45+
leukocyte populations compared to the cohort of patients not requiring
ICU admission.
14.43.3 Limitations
The limitations of this study include the relatively
small sample size and lack of longitudinal testing. The authors also did
not assess whether respiratory comorbidity – such as asthma or chronic
obstructive lung disease – in addition to immunosuppression affected ICU
admission likelihood.
14.43.4 Significance
COVID-19 has already sickened thousands across the globe,
though the severity of these infections is markedly diverse, ranging
from mild symptoms to respiratory failure requiring maximal
intervention. Understanding what clinical, laboratory, and immunologic
factors predict the clinical course of COVID-19 infection permits
frontline providers to distribute limited medical resources more
effectively.
14.43.5 Credit
Review by Andrew Charap as part of a project by students, postdocs and
faculty at the Immunology Institute of the Icahn School of Medicine at
Mount Sinai.
14.44 Serological detection of 2019-nCoV respond to the epidemic: A useful complement to nucleic acid testing
This study showed that both anti-2019-nCov IgM and IgG were detected by
automated chemiluminescent immunoassay in the patients who had been
already confirmed as positive by nucleic acid detection, while single
positivity of IgM or IgG were detected in a very few cases in the other
population including 225 non-COVID-19 cases. In addition to the increase
of anti-2019-nCov IgM 7-12 days after morbidity, the increase of IgG was
detected in three patients with COVID-19 within a very short of time
(0-1 day).
14.44.3 Limitations
The limitation of this study is only
3 confirmed COVID-19 cases were included, so that the relationship
between anti-2019-nCov antibodies and disease progression might not be
clearly defined. Another limitation is that they did not show the course
of 2019-nCov specific antibodies in the cases with positive for COVID-19
but without clinical symptoms.
14.44.4 Significance
The detection of anti-2019-nCov antibodies can be an alternative method
to diagnose and treat COVID-19 more comprehensively by distinguish non
COVID-19 patients. It may be helpful to understand the course of
individual cases with COVID-19 to predict the prognosis if more cases
will be evaluated.
14.44.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.45 Human Kidney is a Target for Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection
Analyzing the eGFR (effective glomerular flow rate) of 85 Covid-19
patients and characterizing tissue damage and viral presence in
post-mortem kidney samples from 6 Covid-19 patients, the authors
conclude that significant damage occurs to the kidney, following
Covid-19 infection. This is in contrast to the SARS infection from the
2003 outbreak. They determine this damage to be more prevalent in
patients older than 60 years old, as determined by analysis of eGFR. H&E
and IHC analysis in 6 Covid-19 patients revealed that damage was in the
tubules, not the glomeruli of the kidneys and suggested that macrophage
accumulation and C5b-9 deposition are key to this process.
14.45.3 Limitations
Severe limitations include that the H&E and IHC samples were performed
on post-mortem samples of unknown age, thus we cannot assess how/if age
correlates with kidney damage, upon Covid-19 infection. Additionally,
eGFR was the only in-vivo measurement. Blood urea nitrogen and
proteinuria are amongst other measurements that could have been obtained
from patient records. An immune panel of the blood was not performed to
assess immune system activation. Additionally, patients are only from
one hospital.
14.45.4 Significance
This report makes clear that kidney damage is prevalent
in Covid-19 patients and should be accounted for.
14.45.5 Credit
Review by Dan Fu Ruan, Evan Cody and Venu Pothula as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine at Mount Sinai.
14.46 COVID-19 early warning score: a multi-parameter screening tool to identify highly suspected patients
The aim of this study was to identify diagnostic or prognostic criteria
which could identify patients with COVID-19 and predict patients who
would go on to develop severe respiratory disease. The authors use EMR
data from individuals taking a COVID-19 test at Zhejiang hospital, China
in late January/Early February. A large number of clinical parameters
were different between individuals with COVID-19 and also between
‘severe’ and ‘non-severe’ infections and the authors combine these into
a multivariate linear model to derive a weighted score, presumably
intended for clinical use.
14.46.3 Limitations
Unfortunately, the paper is lacking a lot of crucial information, making
it impossible to determine the importance or relevance of the findings.
Most importantly, the timings of the clinical measurements are not
described relative to the disease course, so it is unclear if the
differences between ‘severe’ and ‘non-severe’ infections are occurring
before progression to severe disease (which would make them useful
prognostic markers), or after (which would not).
14.46.4 Significance
This paper is one of many retrospective studies coming from hospitals in
China studying individuals with COVID-19. Because of the sparse
description of the study design, this paper offers little new
information. However, studies like this could be very valuable and we
would strongly encourage the authors to revise this manuscript to
include more information about the timeline of clinical measurements in
relation to disease onset and more details of patient outcomes.
14.46.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.47 LY6E impairs coronavirus fusion and confers immune control of viral disease
Screening a cDNA library of >350 human interferon-stimulated genes
for antiviral activity against endemic human coronavirus HCoV-229E
(associated with the common cold), Pfaender S & Mar K et al. identify
lymphocyte antigen 6 complex, locus E (Ly6E) as an inhibitor of cellular
infection of Huh7 cells, a human hepatoma cell line susceptible to
HCoV-229E and other coronaviruses. In a series of consecutive in vitro
experiments including both stable Ly6E overexpression and
CRISPR-Cas9-mediated knockout the authors further demonstrate that Ly6E
reduces cellular infection by various other coronaviruses including
human SARS-CoV and SARS-CoV-2 as well as murine CoV mouse hepatitis
virus (MHV). Their experiments suggest that this effect is dependent on
Ly6E inhibition of CoV strain-specific spike protein-mediated membrane
fusion required for viral cell entry.
To address the function of Ly6E in vivo, hematopoietic stem
cell-specific Ly6E knock-out mice were generated by breeding Ly6Efl/fl
mice (referred to as functional wild-type mice) with transgenic
Vav-iCre mice (offspring referred to as Ly6E HSC ko mice); wild-type
and Ly6E HSC ko mice of both sexes were infected intraperitoneally with
varying doses of the natural murine coronavirus MHV, generally causing a
wide range of diseases in mice including hepatitis, enteritis and
encephalomyelitis. Briefly, compared to wild-type controls, mice lacking
hematopoietic cell-expressed Ly6E were found to present with a more
severe disease phenotype as based on serum ALT levels (prognostic of
liver damage), liver histopathology, and viral titers in the spleen.
Moreover, bulk RNAseq analysis of infected liver and spleen tissues
indicated changes in gene expression pathways related to tissue damage
and antiviral immune responses as well as a reduction of genes
associated with type I IFN response and inflammation. Finally, the
authors report substantial differences in the numbers of hepatic and
splenic APC subsets between wild-type and knockout mice following MHV
infection and show that Ly6E-deficient B cells and to a lesser extent
also DCs are particularly susceptible to MHV infection in vitro.
14.47.3 Limitations
Experiments and data in this study are presented in an overall logical
and coherent fashion; however, some observations and the conclusions
drawn are problematic and should be further addressed & discussed by the
authors. Methodological & formal limitations include relatively low
replicate numbers as well as missing technical replicates for some in
vitro experiments (cf. Fig. legend 1; Fig. legend 2e); the omission
of “outliers” in Fig. legend 2 without an apparent rationale as to why
this approach was chosen; the lack of detection of actual Ly6E protein
levels in Ly6E HSC ko or wild-type mice; and most importantly, missing
information on RNAseq data collection & analysis in the method section
and throughout the paper. A more relevant concern though is that the
interpretation of the experimental data presented and the language used
tend to overrate and at times overgeneralize findings: for example,
while the authors demonstrate statistically significant, Ly6E-mediated
reduction of coronavirus titers in stable cells lines in vitro, it
remains unclear whether a viral titer reduction by one log decade would
be of actual biological relevance in face of high viral titers in
vivo. After high-dose intraperitoneal MHV infection in vivo, early
viral titers in Ly6E HSC knockout vs. wt mice only showed an elevation
in the spleen (~1.5 log decades) but not liver of the ko mice (other
tissue not evaluated), and while ko mice presented with only modestly
increased liver pathology, both male and female ko mice exhibited
significantly higher mortality. Thus, the manuscript tile statement that
“Ly6E … confers immune control of viral disease” is supported by only
limited in vivo data, and gain-of-function experiments (eg. Ly6E
overexpression) were not performed. Of additional note here, tissue
tropism and virulence differ greatly among various MHV strains and
isolates whereas dose, route of infection, age, genetic background and
sex of the mice used may additionally affect disease outcome and
phenotype (cf. Taguchi F & Hirai-Yuki A,
https://doi.org/10.3389/fmicb.2012.00068; Kanolkhar A et al,
https://jvi.asm.org/content/
83/18/9258). Observations
attributed to hematopoietic stem cell-specific Ly6E deletion could
therefore be influenced by the different genetic backgrounds of floxed
and cre mice used, and although it appears that littermates wt and ko
littermates were used in the experiments, the potentially decisive
impact of strain differences should at least have been discussed. Along
these lines, it should also be taken into account that the majority of
human coronaviruses cause respiratory symptoms, which follow a different
clinical course engaging other primary cellular mediators than the
hepatotropic murine MHV disease studied here. It therefore remains
highly speculative how the findings reported in this study will
translate to human disease and it would therefore be important to test
other routes of MHV infection and doses that have been described to
produce a more comparable phenotype to human coronavirus disease (cf.
Kanolkhar A et al, https://jvi.asm.org/content/
83/18/9258). Another
important shortcoming of this study is the lack of any information on
functional deficits or changes in Ly6E-deficient immune cells and how
this might relate to the phenotype observed. Overall, the in vitro
experiments are more convincing than the in vivo studies which appear
somewhat limited.
14.47.4 Significance
Despite some shortcomings, the experiments performed in this study
suggest a novel and somewhat unexpected role of Ly6E in the protection
against coronaviruses across species. These findings are of relevance
and should be further explored in ongoing research on potential
coronavirus therapies. Yet an important caveat pertains to the authors’
suggestion that “therapeutic mimicking of Ly6E action” may constitute a
first line of defense against novel coronaviruses since their own prior
work demonstrated that Ly6E can enhance rather than curtail infection
with influenza A and other viruses.
14.47.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.48 A preliminary study on serological assay for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in 238 admitted hospital patients
While RT-PCR is being used currently to routinely diagnose infection
with SARS-CoV-2, there are significant limitations to the use of a
nucleic acid test that lead to a high false-negative rate. This
article describes ELISAs that can measure IgM and IgG antibodies
against the N protein of SARS-CoV-2 to test samples from 238
patients (153 positive by RT-PCR and 85 negative by RT-PCR) at
different times after symptom onset. The positivity rate of the IgM
and/or IgG ELISAs was greater than that of the RT-PCR (81.5%
compared to 64.3%) with similar positive rates in the confirmed and
suspected cases (83% and 78.8%, respectively), suggesting that many
of the suspected but RT-PCR-negative cases were also infected. The
authors also found that the ELISAs have higher positive rates later
after symptom onset while RT-PCR is more effective as a diagnostic
test early during the infection.
14.48.3 Limitations
I cannot identify any limitations to this study.
14.48.4 Significance
The authors make a strong case for using a combination of ELISA and
RT-PCR for diagnosis of infection with SARS-CoV-2, especially
considering the dynamics of positivity rates of RT-PCR and ELISA.
Fewer false-negative diagnoses would improve infection control and
patient management.
14.48.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.49 Monoclonal antibodies for the S2 subunit of spike of SARS-CoV cross-react with the newly-emerged SARS-CoV-2
Whole genome sequencing-based comparisons of the 2003
Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) and the 2019
SARS-CoV-2 revealed conserved receptor binding domain (RBD) and host
cell receptor, angiotensin-converting enzyme 2 (ACE2). In line with
this, the authors tested cross-reactivity of murine monoclonal
antibodies (mAbs) previously generated against the SARS-CoV spike (S)
glycoprotein involved in viral entry. One of the screened mAb, 1A9, was
able to bind and cross-neutralize multiple strains of SARS-CoV, as well
as, detect the S protein in SARS-CoV-2-infected cells. mAb 1A9 was
generated using an immunogenic fragment in the S2 subunit of SARS-CoV
and binds through a novel epitope within the S2 subunit at amino acids
1111-1130. It is important to note that CD8+ T lymphocyte epitopes
overlap with these residues, suggesting that S2 subunit could be
involved in inducing both, humoral and cell-mediated immunity.
14.49.3 Limitations
The authors used previously generated mouse mAbs
against the S protein in SARS-CoV expressed in mammalian cell line.
Future experimental validation using COVID-19 patient samples is needed
to validate these findings. In addition, the results of these studies
are predominantly based on in vitro experiments and so, evaluating the
effects of the mAb 1A9 in an animal model infected with this virus will
help us better understand the host immune responses in COVID-19 and
potential therapeutic vaccines.
14.49.4 Significance
This study identified mAbs that recognize the new
coronavirus, SARS-Cov-2. These cross-reactive mAbs will help in
developing diagnostic assays for COVID-19.
14.49.5 Credit
This review was undertaken by Tamar Plitt and Katherine Lindblad as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.50 Mortality of COVID-19 is Associated with Cellular Immune Function Compared to Immune Function in Chinese Han Population
Retrospective study of the clinical characteristics of 752
patients infected with COVID-19 at Chinese PLA General Hospital, Peking
Union Medical College Hospital, and affiliated hospitals at Shanghai
University of medicine & Health Sciences. This study is the first one
that compares PB from healthy controls from the same regions in Shanghai
and Beijing, and infected COVID-19 patients to standardize a reference
range of WBCs of people at high risk.
14.50.3 Limitations
Lower levels of leukocyte counts -B cells, CD4 and CD8 T
cells- correlated with mortality (WBCs are significantly lower in severe
or critical UCI patients vs mild ones). Based on 14,117 normal controls
in Chinese Han population (ranging in age from 18-86) it is recommended
that reference ranges of people at high risk of COVID-19 infection are
CD3+ lymphocytes below 900 cells/mm3, CD4+ lymphocytes below 500
cells/mm3, and CD8+ lymphocytes below 300 cells/mm3. Importantly, this
study also reported that the levels of D-dimer, C-reactive protein and
IL-6 were elevated in COVID-19 pts., indicating clot formation, severe
inflammation and cytokine storm.
14.50.4 Significance
This study sets a threshold to identify patients at risk
by analyzing their levels of leukocytes, which is an easy and fast
approach to stratify individuals that require hospitalization. Although
the study is limited (only counts of WBC are analyzed and not its
profile) the data is solid and statistically robust to correlate levels
of lymphopenia with mortality.
14.50.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.51 Retrospective Analysis of Clinical Features in 101 Death Cases with COVID-19
This is a retrospective study involving 101 death cases with COVID-19 in
Wuhan Jinyintan Hospital. The aim was to describe clinical,
epidemiological and laboratory features of fatal cases in order to
identify the possible primary mortality causes related to COVID-19.
Among 101 death cases, 56.44% were confirmed by RT-PCR and 43.6% by
clinical diagnostics. Males dominated the number of deaths and the
average age was 65.46 years. All patients died of respiratory failure
and multiple organs failure, except one (acute coronary syndrome). The
predominant comorbidities were hypertension (42.57%) and diabetes
(22.77%). 25.74% of the patients presented more than two underlying
diseases. 82% of patients presented myocardial enzymes abnormalities at
admission and further increase in myocardial damage indicators with
disease progression: patients with elevated Troponin I progressed faster
to death. Alterations in coagulation were also detected. Indicators of
liver and kidney damage increased 48 hours before death. The authors
studied the deceased patients’ blood type and presented the following
results: type A (44.44%), type B (29.29%), type AB (8.08%) and type O
(18.19%), which is inconsistent with the distribution in Han population
in Wuhan.
Clinical analysis showed that the most common symptom was fever (91.9%),
followed by cough and dyspnea. The medium time from onset of symptoms to
acute respiratory distress syndrome (ARDS) development was 12 days.
Unlike SARS, only 2 patients with COVID-19 had diarrhea. 98% presented
abnormal lung imaging at admission and most had double-lung
abnormalities. Related to the laboratorial findings some inflammatory
indicators gradually increased during the disease progression, such as
IL-6 secretion in the circulation, procalcitonin (PCT) and C-reactive
protein (CRP), while platelets numbers decreased. The authors also
reported an initial lymphopenia that was followed by an increase in the
lymphocytes numbers. Neutrophil count increased with disease
progression.
The patients received different treatments such as antiviral drugs
(60.40%), glucocorticoids, thymosin and immunoglobulins. All patients
received antibiotic treatment and some received antifungal drugs. All
patients received oxygen therapy (invasive or non-invasive ones).
14.51.3 Limitations
This study involves just fatal patients, lacking comparisons with other
groups of patients e.g. patients that recovered from COVID-19. The
authors didn’t discuss the different approaches used for treatments and
how these may affect the several parameters measured. The possible
relationship between the increase of inflammatory indicators and
morbidities of COVID-19 are not discussed.
14.51.4 Significance
This study has the largest cohort of fatal cases reported so far. The
authors show that COVID-19 causes fatal respiratory distress syndrome
and multiple organ failure. This study highlights prevalent myocardial
damage and indicates that cardiac function of COVID-19 patients should
be carefully monitored. The data suggest that Troponin I should be
further investigated as an early indicator of patients with high risk of
accelerated health deterioration. Secondary bacterial and fungal
infections were frequent in critically ill patients and these need to be
carefully monitored in severe COVID-19 patients. Differences in blood
type distribution were observed, suggesting that type A is detrimental
while type O is protective – but further studies are needed to confirm
these findings and elucidate if blood type influences infection or
disease severity. Several inflammatory indicators (neutrophils, PCT, CRP
and IL-6, D-dimer) increased according to disease severity and should be
assessed as biomarkers and to better understand the biology of
progression to severe disease.
14.51.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.52 Relationship between the ABO Blood Group and the COVID-19 Susceptibility
These authors compared the ABO blood group of 2,173 patients with
RT-PCR-confirmed COVID-19 from hospitals in Wuhan and Shenzhen with the
ABO blood group distribution in unaffected people in the same cities
from previous studies (2015 and 2010 for Wuhan and Shenzhen,
respectively). They found that people with blood group A are
statistically over-represented in the number of those infected and who
succumb to death while those with blood group O are statistically
underrepresented with no influence of age or sex.
14.52.3 Limitations
This study compares patients with COVID-19 to the general population
but relies on data published 5 and 10 years ago for the control. The
mechanisms that the authors propose may underlie the differences they
observed require further study.
14.52.4 Significance
Risk stratification based on blood group may be beneficial for
patients and also healthcare workers in infection control. Additionally,
investigating the mechanism behind these findings could lead to better
developing prophylactic and therapeutic targets for COVID-19.
14.52.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.53 The inhaled corticosteroid ciclesonide blocks coronavirus RNA replication by targeting viral NSP15
This study reconsiders the use of inhaled corticosteroids in the
treatment of pneumonia by coronavirus. Corticosteroids were associated
with increased mortality for SARS in 2003 and for MERS in 2013, probably
due to that fact that systemic corticosteroids suppress the innate
immune system, resulting in increased viral replication. However, some
steroid compounds might block coronavirus replication. The authors
screened steroids from a chemical library and assessed the viral growth
suppression and drug cytotoxicity. Ciclesonide demonstrated low
cytotoxicity and potent suppression of MERS-CoV viral growth. The
commonly used systemic steroids cortisone, prednisolone and
dexamethasone did not suppress viral growth, nor did the commonly used
inhaled steroid fluticasone. To identify the drug target of virus
replication, the authors conducted 11 consecutive MERS-CoV passages in
the presence of ciclesonide or mometasone, and they could generate a
mutant virus that developed resistance to ciclesonide, but not to
mometasone. Afterwards, they performed next-generation sequencing and
identified an amino acid substitution in nonstructural protein 15
(NSP15) as the predicted mechanism for viral resistance to ciclesonide.
The authors were able to successfully generate a recombinant virus
carrying that amino acid substitution, which overcome the antiviral
effect of ciclesonide, suggesting that ciclosenide interacts with NSP15.
The mutant virus was inhibited by mometasone, suggesting that the
antiviral target of mometasone is different from that of ciclesonide.
Lastly, the effects of ciclesonide and mometason on suppressing the
replication of SARS-CoV-2 were evaluated. Both compounds were found to
suppress viral replication with a similar efficacy to lopinavir.
14.53.3 Limitations
Most of the experiments, including the identification of the mutation in
NSP15 were conducted with MERS-CoV. This is not the closest related
virus to SARS-CoV-2, as that would be SARS-CoV. Thus, to repeat the
initial experiments with SARS-CoV, or preferably SARS-CoV-2, is
essential. The manuscript should address this and, therefore, it will
require considerable editing for organization and clarity. Also, in
terms of cell immunogenic epitopes, while SARS-CoV-2 spike protein
contains several predicted B and T cell immunogenic epitopes that are
shared with other coronaviruses, some studies have shown critical
differences between MERS-CoV, SARS-CoV and SARS-CoV-2. A main criticism
is that the authors only used VeroE6/TMPRSS2 cells to gauge the direct
cytotoxic effects of viral replication. To evaluate this in other cell
lines, including human airway epithelial cells, is crucial, as the
infectivity of coronavirus strains greatly varies in different cell
lines,
14.53.4 Significance
Nevertheless, these findings encourage evaluating ciclesonide and
mometasone as better options for patients with COVID-19 in need of
inhaled steroids, especially as an alternative to other corticosteroids
that have been shown to increase viral replication in vitro. This should
be evaluated in future clinical studies.
14.53.5 Credit
This review was undertaken by Alvaro Moreira, MD as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.54 A human monoclonal antibody blocking SARS-CoV-2 infection **
The authors reported a human monoclonal
antibody that neutralizes SARS-CoV-2 and SARS-Cov which belong to same
family of corona viruses. For identifying mAbs, supernatants of a
collection of 51 hybridomas raised against the spike protein of SARS-CoV
(SARS-S) were screened by ELISA for cross-reactivity against the spike
protein of SARS-CoN2 (SARS2-S). Hybridomas were derived from immunized
transgenic H2L2 mice (chimeric for fully human VH-VL and rat constant
region). Four SARS-S hybridomas displayed cross-reactivity with SARS2-S,
one of which (47D11) exhibited cross-neutralizing activity for SARS-S
and SARS2-S pseudotyped VSV infection. A recombinant, fully human IgG1
isotype antibody was generated and used for further characterization.
The humanized 47D11 antibody inhibited infection of VeroE6 cells with
SARS-CoV and SARS-CoV-2 with IC50 values of 0.19 and 0.57 μg/ml
respectively. 47D11 mAb bound a conserved epitope on the spike receptor
binding domain (RBD) explaining its ability to cross-neutralize SARS-CoV
and SARS-CoV-2. 47D11 was shown to target the S1B RBD of SARS-S and
SARS2-S with similar affinities. Interestingly, binding of 47D11 to
SARS-S1B and SARS2-S1B did not interfere with S1B binding to ACE2
receptor-expressing cells assayed by flow cytometry.
14.54.3 Limitations
These results show that the human 47D11 antibody
neutralizes SARS-CoV and SARS-Cov2 infectivity via an as yet unknown
mechanism that is different from receptor binding interference.
Alternative mechanisms were proposed but these as yet remain to be
tested in the context of SARS-CoV2. From a therapeutic standpoint and in
the absence of in vivo data, it is unclear whether the 47D11 ab can
alter the course of infection in an infected host through virus
clearance or protect an uninfected host that is exposed to the virus.
There is a precedent for the latter possibility as it relates to
SARS-CoV that was cited by the authors and could turn out to be true for
SARS-CoV2.
14.54.4 Significance
This study enabled the identification of novel neutralizing
antibody against COV-that could potentially be used as first line of
treatment in the near future to reduce the viral load and adverse
effects in infected patients. In addition, neutralizing antibodies such
as 47D11 represent promising reagents for developing
antigen-antibody-based detection test kits and assays.
14.54.5 Credit
This review was edited by K. Alexandropoulos as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
Heat inactivation of serum interferes with the immunoanalysis of
antibodies to SARS-CoV-2
The use of heat inactivation to neutralize pathogens in serum samples
collected from suspected COVID-19 patients reduces the sensitivity of a
fluorescent immunochromatographic assay to detect anti-SARS-CoV-2 IgM
and IgG.
Major findings
Coronaviruses can be killed by heat inactivation, and this is an
important safety precaution in laboratory manipulation of clinical
samples. However, the effect of this step on downstream
SARS-CoV-2-specific serum antibody assays has not been examined. The
authors tested the effect of heat inactivation (56 deg C for 30 minutes)
versus no heat inactivation on a fluorescence immunochromatography
assay. Heat inactivation reduced all IgM measurements by an average of
54% and most IgG measurements (22/36 samples, average reduction of 50%),
consistent with the lower thermal stability of IgM than that of IgG.
Heat inactivation caused a subset of IgM but not IgG readings to fall
below a specified positivity threshold.
Limitations
Limitations included the use of only one type of assay for testing heat
inactivated vs non-inactivated sera, and the use of the same baseline
for heat inactivated and non-inactivated sera. The results indicate that
heat inactivation affects the quantification of SARS-CoV-2-antibody
response, specially IgM, but still allows to distinguish positive
specific IgG. Therefore, the effect of heat inactivation should be
studied when designing assays that quantitatively associate
immunoglobulin levels (especially IgM) to immune state.
Review by Andrew M. Leader as part of a project by students, postdocs
and faculty at the Immunology Institute of the Icahn school of medicine,
Mount Sinai.
14.55 Immune phenotyping based on neutrophil-to-lymphocyte ratio and IgG predicts disease severity and outcome for patients with COVID-19
In a cohort of 222 patients, anti-SARS-CoV-2 IgM and IgG levels were
analyzed during acute and convalescent phases (up to day 35) and
correlated to the diseases’ severity. The same was done with
neutrophil-to-lymphocyte ratio. High IgG levels and high
neutrophil-to-lymphocyte ratio in convalescence were both independently
associated to the severity of the disease. The simultaneous occurrence
of both of these laboratory findings correlated even stronger to the
diseases’ severity.
Severe cases with high neutrophil-to-lymphocyte ratios had clearly
higher levels of IL-6. The authors propose that a robust IgG response
leads to immune-mediated tissue damage, thus explaining the worse
outcome in patients with overexuberant antibody response.
14.55.3 Limitations
A main criticism is that the criteria for stratifying patients in severe
vs. non-severe are not described. The only reference related to this is
the difference between the percentage of patients who needed mechanical
ventilation, which was greater in patients with both high IgG levels and
high neutrophil-to-lymphocyte ratio. No patient with both low IgG levels
and low neutrophil-to-lymphocyte ratio was treated with mechanical
ventilation.
The proposed correlation of severity with IL-2 and IL-10 levels is not
very strong.
Furthermore, although mostly ignored in the paper’s discussion, one of
the most interesting findings is that an early increase in
anti-SARS-CoV-2 IgM levels also seems to correlate with severe disease.
However, as only median values are shown for antibody kinetics curves,
the extent of variation in acute phase cannot be assessed.
14.55.4 Significance
Anti-SARS-CoV-2 IgG levels and with neutrophil-to-lymphocyte ratio
predict severity of COVID-19 independently of each other. An additive
predictive value of both variables is noticeable. Importantly, an
early-on increase in anti-SARS-CoV-2 IgM levels also seem to predict
outcome.
14.55.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.56 Reinfection could not occur in SARS-2 CoV-2 infected rhesus macaques
This study addresses the issue or acquired immunity after a primary
COVID-19 infection in rhesus monkeys. Four Chinese rhesus macaques were
intratracheally infected with SARS-CoV-2 and two out of the four were
re-infected at 28 days post initial infection (dpi) with the same viral
dose after confirming the recovery by the absence of clinical symptoms,
radiological abnormalities and viral detection (2 negative RT-PCR
tests). While the initial infection led the viral loads in nasal and
pharyngeal swabs that reach approximately 6.5 log10 RNA copies/ml at 3
dpi in all four monkeys, viral loads in the swabs tested negative after
reinfection in the two reinfected monkeys. In addition, the necropsies
from a monkey (M1) at 7 days after primary infection, and another monkey
(M3) at 5 days post reinfection, revealed the histopathological damages
and viral replication in the examined tissues from M1, while no viral
replication as well as no histological damages were detected in the
tissues from M3. Furthermore, sera from three monkeys at 21 and 28 dpi
exhibited neutralizing activity against SARS-CoV-2 in vitro, suggesting
the production of protective neutralizing antibodies in these monkeys.
Overall, this study indicates that primary infection with SARS-CoV-2 may
protect from subsequent exposure to the same virus.
14.56.3 Limitations
In human, virus has been detected by nasopharyngeal swabs until 9 to 15
days after the onset of symptoms. In the infected monkeys in this study,
virus were detected from day 1 after the infection, declining to
undetectable level by day 15 post infection. It may suggest that there
is a faster viral clearance mechanism in monkeys, therefore the
conclusions of reinfection protection for humans need to be carefully
considered. In addition, only two monkeys were re-infected in this study
and the clinical signs of these monkeys were not similar: M3 did not
show weight loss and M4 showed relatively higher fever on the day of
infection and the day of re-challenge.
14.56.4 Significance
This study showed clear viral clearance and no indications of relapse or
viremia after a secondary infection with SARS-CoV-2 in a Chinese rhesus
macaque model. These results support the idea that patients with full
recovery (two negative RT-PCR results) may also be protected from
secondary SARS-CoV-2 infection. Recovered patients may be able to
reintegrate to normal public life and provide protective serum perhaps
even if having had a mild infection. The results are also encouraging
for successful vaccine development against SARS-CoV-2.
14.56.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.57 A highly conserved cryptic epitope in the receptor-binding domains of SARS-CoV-2 and SARS-CoV
Given the sequence similarity of the surface spike glycoprotein (S) of
SARS-CoV-2 and SAR-CoV, Yuan et al. (2020) propose that neutralizing
antibodies isolated from convalescent SARS-CoV patients may offer
insight into cross-reactive antibodies targeting SARS-CoV-2. In
particular, they find that the receptor-binding domain (RBD) of
SARS-CoV-2 S protein shares 86% sequence similarity with the RBD of
SARS-CoV S protein that binds to the CR3022 neutralizing antibody.
CR3022 also displays increased affinity for the “up” conformation of the
SARS-CoV-2 S protein compared to the “down” conformation as it does for
the SARS-CoV S protein. Therefore, the authors propose that this
cross-reactive antibody may confer some degree of protection in vivo
even if it fails to neutralize in vitro.
14.57.3 Limitations
Although the authors offer a logical rationale for
identifying cross-reactive neutralizing antibodies derived from
SARS-CoV, their study using only CR3022 failed to demonstrate whether
this approach will be successful. After all, CR3022 failed to neutralize
in vitro despite the binding affinity to a similar epitope on
SARS-CoV-2. They would benefit from testing more candidates and using an
in vivo model to demonstrate their claim that protection may be
possible in the absence neutralization if combinations are used in
vivo.
14.57.4 Significance
The ability to make use of previously characterized
neutralizing antibodies for conserved epitopes can expedite drug design
and treatment options.
14.57.5 Credit
This review was undertaken by Dan Fu Ruan, Evan Cody and Venu Pothula as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.58 Highly accurate and sensitive diagnostic detection of SARS-CoV-2 by digital PCR
The authors present a digital PCR (dPCR) diagnostic test for
SARS-CoV-2 infection. In 103 individuals that were confirmed in a
follow-up to be infected, the standard qPCR test had a positivity
rate of 28.2% while the dPCR test detected 87.4% of the infections
by detecting an additional 61 positive cases. The authors also
tested samples from close contacts (early in infection stage) and
convalescing individuals (late in infection stage) and were able to
detect SARS-CoV-2 nucleic acid in many more samples using dPCR
compared to qPCR.
14.58.3 Limitations
I did not detect limitations.
14.58.4 Significance
The authors make a strong case for the need for a highly sensitive
and accurate confirmatory method for diagnosing COVID-19 during this
outbreak and present a potential addition to the diagnostic arsenal.
They propose a dPCR test that they present has a dramatically lower
false negative rate than the standard RT-qPCR tests and can be
especially beneficial in people with low viral load, whether they
are in the earlier or later stages of infection.
14.58.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.59 SARS-CoV-2 invades host cells via a novel route: CD147-spike protein
The authors propose a novel mechanism of SARS-CoV-2 viral entry through
the interaction of the viral spike protein (SP) and the immunoglobulin
superfamily protein CD147 (also known as Basigin). Using an in-house
developed humanized antibody against CD147 (maplazumab), they show that
blocking CD147 decreases viral replication in Vero E6 cells. Using
surface plasmon resonance (SPR), ELISA, and Co-IP assays, they show that
the spike protein of SARS-CoV-2 directly interacts with CD147. Lastly,
they utilize immune-election microscopy to show spike protein and CD147
localize to viral inclusion bodies of Vero E6 cells.
14.59.3 Limitations
The authors claim that an anti-CD147 antibody (Meplazumab) inhibits
SARS-CoV-2 replication by testing cell growth and viral load in cells
infected with SARS-CoV-2, however there are key pieces of this
experiment that are missing. First, the authors fail to use a
non-specific antibody control. Second, the authors claim that viral
replication is inhibited, and that they test this by qPCR, however this
data is not shown. To further prove specificity, the authors should
introduce CD147 to non-susceptible cells and show that they become
permissive.
The authors claim that there is a direct interaction between CD147 and
SP through SPR, ELISA, and Co-IP, and this data seems generally
convincing. The electron microscopy provides further correlative
evidence that SARS-CoV-2 may interact with CD147 as they are both found
in the same viral inclusion body. A quantification of this data would
make the findings more robust.
Finally, the data in this paper lacks replicates, error bars, and
statistics to show that the data are reproducible and statistically
significant.
14.59.4 Significance
It has been shown in various studies that SARS-CoV-2 binds to the cell
surface protein ACE2 for cell entry, yet ACE2 is highly expressed in
heart, kidney, and intestinal cells, raising the concern that blocking
ACE2 would result in harmful side effects (2233) CD147 on the other hand
is highly expressed in various tumor types, inflamed tissues, and
pathogen infected cells, suggesting that the inhibition of CD147 would
not result in major side effects (2234, 2235) The research in this paper has
resulted in an ongoing clinical trail in China to test the safety and
efficacy of anti-CD147 Meplazumab to treat COVID-19. (ClinicalTrails.gov
identifier NCT04275245).
14.59.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.60 Blood single cell immune profiling reveals that interferon-MAPK pathway mediated adaptive immune response for COVID-19
The authors performed single-cell RNA sequencing (scRNAseq) of
peripheral blood mononuclear cells isolated from whole blood samples of
COVID-19 patients (n=10). Data was compared to scRNAseq of samples
collected from patients with influenza A (n=1), acute pharyngitis (n=1),
and cerebral infarction (n=1), as well as, three healthy controls.
COVID-19 patients were categorized into those with moderate (n=6),
severe (n=1), critical (n=1), and cured (n=2) disease. Analysis across
all COVID-19 disease levels revealed 56 different cellular subtypes,
among 17 immune cell types; comparisons between each category to the
normal controls revealed increased proportions of CD1c+ dendritic
cells, CD8+ CTLs, and plasmacytoid dendritic cells and a decrease in
proportions of B cells and CD4+ T cells.
TCR sequencing revealed that greater clonality is associated with
milder COVID-19 disease; BCR sequencing revealed that COVID-19
patients have circulating antibodies against known viral antigens,
including EBV, HIV, influenza A, and other RNA viruses. This may suggest
that the immune response to SARS-CoV-2 infection elicits production of
antibodies against known RNA viruses.
Excluding enriched pathways shared by COVID-19 patients and patients
with other conditions (influenza A, acute pharyngitis, and cerebral
infarction), the authors identified the interferon-MAPK signaling
pathway as a major response to SARS-CoV-2 infection. The authors
performed quantitative real-time reverse transcriptase polymerase chain
reaction (RT-PCR) for interferon-MAPK signaling genes: IRF27, BST2,
and FOS. These samples were collected from a separate cohort of
COVID-19 patients (critical, n=3; severe, n=3; moderate, n=19; mild,
n=3; and cured, n=10; and healthy controls, n=5). Notably, consistent
with the original scRNAseq data, FOS showed up-regulation in COVID-19
patients and down-regulation in cured patients. The authors propose
that FOS may be a candidate marker gene for curative COVID-19
disease.
14.60.3 Limitations
The sample size of this study is limited. To further delineate
differences in the immune profile of peripheral blood of COVID-19
patients, a greater sample size is needed, and longitudinal samples are
needed, as well. A better understanding of the immunological
interactions in cured patients, for example, would require a profile
before and after improvement.
Moreover, the conclusions drawn from this scRNAseq study point to
potential autoimmunity and immune deficiency to distinguish different
severities of COVID-19 disease. However, this requires an expanded
number of samples and a more robust organization of specific immune cell
subtypes that can be compared across different patients. Importantly,
this criterion is likely needed to ensure greater specificity in
identifying markers for COVID-19 infection and subsequent immune
response.
14.60.4 Significance
At the single-cell level, COVID-19 disease has been characterized in the
lung, but a greater understanding of systemic immunological responses is
furthered in this study. Type I interferon is an important signaling
molecule for the anti-viral response. The identification of the
interferon-MAPK signaling pathway and the differential expression of
MAPK regulators between patients of differing COVID-19 severity and
compared to cured patients may underscore the importance of either
immune deficiency or autoimmunity in COVID-19 disease.
14.60.5 Credit
This review was undertaken by Matthew D. Park as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.61 Cross-reactive antibody response between SARS-CoV-2 and SARS-CoV infection.
The authors explore the antigenic differences between SARS-CoV-2 and
SARS-CoV by analyzing plasma samples from SARS-CoV-2 (n = 15) and
SARS-CoV (n = 7) patients. Cross-reactivity in antibody binding to the
spike protein between SARS-CoV-2 and SARS-CoV was found to be common,
mostly targeting non-RBD regions in plasma from SARS-CoV-2 patients.
Only one SARS-CoV-2 plasma sample was able to cross-neutralize SARS-CoV,
with low neutralization activity. No cross-neutralization response was
detected in plasma from SARS-CoV patients.
To further investigate the cross-reactivity of antibody responses to
SARS-CoV-2 and SARS-CoV, the authors analyzed the antibody response of
plasma collected from mice infected or immunized with SARS-CoV-2 or
SARS-CoV (n = 5 or 6 per group). Plasma from mice immunized with
SARS-CoV-2 displayed cross-reactive responses to SARS-CoV S ectodomain
and, to a lesser extent, SARS-CoV RBD. Similarly, plasma from mice
immunized with SARS-CoV displayed cross-reactive responses to SARS-CoV-2
S ectodomain. Cross-neutralization activity was not detected in any of
the mouse plasma samples.
14.61.3 Limitations
The size of each patient cohort is insufficient to accurately determine
the frequency of cross-reactivity and cross-neutralization in the
current SARS-CoV-2 pandemic. Recruitment of additional patients from a
larger range of geographical regions and time points would also enable
exploration into the effect of the genetic diversity and evolution of
the SARS-CoV-2 virus on cross-reactivity. This work would also benefit
from the mapping of specific epitopes for each sample. Future studies
may determine whether the non-neutralizing antibody responses can confer
in vitro protection or lead to antibody-dependent disease enhancement.
14.61.4 Significance
The cross-reactive antibody responses to S protein in the majority of
SARS-CoV-2 patients is an important consideration for development of
serological assays and vaccine development during the current outbreak.
The limited extent of cross-neutralization demonstrated in this study
indicates that vaccinating to cross-reactive conserved epitopes may have
limited efficacy, presenting a key concern for the development of a more
universal coronavirus vaccine to address the global health risk of novel
coronavirus outbreaks.
14.61.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.62 The feasibility of convalescent plasma therapy in severe COVID-19 patients: a pilot study
This is the first report to date of convalescent plasma therapy as a
therapeutic against COVID-19 disease. This is a feasibility pilot study.
The authors report the administration and clinical benefit of 200 mL of
convalescent plasma (CP) (1:640 titer) derived from recently cured
donors (CP selected among 40 donors based on high neutralizing titer and
ABO compatibility) to 10 severe COVID-19 patients with confirmed
viremia. The primary endpoint was the safety of CP transfusion. The
secondary endpoint were clinical signs of improvement based on symptoms
and laboratory parameters.
The authors reported use of methylene blue photochemistry to inactivate
any potential residual virus in the plasma samples, without compromising
neutralizing antibodies, and no virus was detected before transfusion.
The authors report the following:
No adverse events were observed in all patients, except 1 patient
who exhibited transient facial red spotting.
All patients showed significant improvement in or complete
disappearance of clinical symptoms, including fever, cough,
shortness of breath, and chest pain after 3 days of CP therapy.
Reduction of pulmonary lesions revealed by chest CT.
Elevation of lymphocyte counts in patients with lymphocytopenia.
Increase in SaO2 in all patients, indicative of recuperating lung
function.
Resolution of SARS-CoV-2 viremia in 7 patients and increase in
neutralizing antibody titers in 5 patients. Persistence of
neutralizing antibody levels in 4 patients.
14.62.3 Limitations
It is important to note that most recipients had high neutralization
titers of antibodies before plasma transfusion and even without
transfusion it would be expected to see an increase in neutralizing
antibodies over time. In addition to the small sample set number (n=10),
there are additional limitations to this pilot study:
All patients received concurrent therapy, in addition to the CP
transfusion. Therefore, it is unclear whether a combinatorial or
synergistic effect between these standards of care and CP
transfusion contributed to the clearance of viremia and improvement
of symptoms in these COVID-19 patients.
The kinetics of viral clearance was not investigated, with respect
to the administration of CP transfusion. So, the definitive impact
of CP transfusion on immune dynamics and subsequent viral load is
not well defined.
Comparison with a small historical control group is not ideal.
14.62.4 Significance
For the first time, a pilot study provides promising results involving
the use of convalescent plasma from cured COVID-19 patients to treat
others with more severe disease. The authors report that the
administration of a single, high-dose of neutralizing antibodies is
safe. In addition, there were encouraging results with regards to the
reduction of viral load and improvement of clinical outcomes. It is,
therefore, necessary to expand this type of study with more
participants, in order to determine optimal dose and treatment kinetics.
It is important to note that CP has been studied to treat H1N1
influenza, SARS-CoV-1, and MERS-CoV, although it has not been proven to
be effective in treating these infections.
14.62.5 Credit
Review by Matthew D. Park and revised by Alice O. Kamphorst and Maria A. Curotto de Lafaille as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.63 Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open label non-randomized clinical trial
This study was a single-arm, open label clinical trial with
600 mg hydroxychloroquine (HCQ) in the treatment arm (n = 20). Patients
who refused participation or patients from another center not treated
with HCQ were included as negative controls (n = 16). Among the patients
in the treatment arm, 6 received concomitant azithromycin to prevent
superimposed bacterial infection. The primary endpoint was respiratory
viral loads on day 6 post enrollment, measured by nasopharyngeal swab
followed by real-time reverse transcription-PCR.
HCQ alone was able to significantly reduce viral loads by day 6 (n =
8/14, 57.1% complete clearance, p < 0.001); azithromycin appears to
be synergistic with HCQ, as 6/6 patients receiving combined treatment
had complete viral clearance (p < 0.001).
14.63.3 Limitations
Despite what is outlined above, this study has a number
of limitations that must be considered. First, there were originally n =
26 patients in the treatment arm, with 6 lost to follow up for the
following reasons: 3 transferred to ICU, 1 discharge, 1
self-discontinued treatment d/t side effects, and 1 patient expired.
Total length of clinical follow up was 14 days, but the data beyond day
6 post-inclusion are not shown.
Strikingly, in supplementary table 1, results of the real-time RT-PCR
are listed for the control and treatment arms from D0 – D6. However, the
data are not reported in a standard way, with a mix of broadly positive
or negative result delineation with Ct (cycle threshold) values, the
standard output of real time PCR. It is impossible to compare what is
defined as a positive value between the patients in the control and
treatment arms without a standardized threshold for a positive test.
Further, the starting viral loads reported at D0 in the groups receiving
HCQ or HCQ + azithromycin were significantly different (ct of 25.3 vs
26.8 respectively), which could explain in part the differences observed
in the response to treatment between 2 groups. Finally, patients in the
control arm from outside the primary medical center in this study
(Marseille) did not actually have samples tested by PCR daily. Instead,
positive test results from every other day were extrapolated to mean
positive results on the day before and after testing as well (Table 2,
footnote a).
Taken together, the results of this study suggest that HCQ represents a
promising treatment avenue for COVID-19 patients. However, the limited
size of the trial, and the way in which the results were reported does
not allow for other medical centers to extrapolate a positive or
negative result in the treatment of their own patients with HCQ +/-
azithromycin. Further larger randomized clinical trials will be required
to ascertain the efficacy of HCQ +/- azithromycin in the treatment of
COVID-19.
14.63.4 Significance
Chloroquine is thought to inhibit viral infection,
including SARS-Cov-2, by increasing pH within endosomes and lysosomes,
altering the biochemical conditions required for viral fusion (806, 2240).
However, chloroquine also has immuno-modulatory effects that I think may
play a role. Chloroquine has been shown to increase CTLA-4 expression at
the cell surface by decreasing its degradation in the endo-lysosome
pathway; AP-1 traffics the cytoplasmic tail of CTLA-4 to lysosomes, but
in conditions of increased pH, the protein machinery required for
degradation is less functional (2241). As such, more CTLA-4 remains in
endosomes and is trafficked back to the cell surface. It is possible
that this may also contribute to patient recovery via reduction of
cytokine storm, in addition to the direct anti-viral effects of HCQ.
14.63.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.64 Recapitulation of SARS-CoV-2 Infection and Cholangiocyte Damage with Human Liver Organoids
Used human liver ductal organoids to determine ACE2+ cholangiocytes
in healthy liver (2.92% of all cells) are infectable and support
SARS-CoV-2 viral replication.
Plaque-purified SARS-CoV-2 viral infection disrupted organoid
barrier and bile transporting functions of cholangiocytes through
dysregulation of genes involved in tight junction formation (CLDN1)
and bile acid transportation (ASBT and CFTR).
14.64.3 Limitations:
Unclear if liver damage observed in patients due to direct
cholangiocyte infection or due to secondary immune/cytokine effects.
This study argues for direct damage as it lacks immune contexture;
but further studies needed with autopsy samples or organoid-immune
cell co-culture to conclude strongly.
Would be important to measure cholangiocyte-intrinsic anti-viral
response and alarmins secreted upon infection, and furthermore study
tropism of various immune cells to conditioned media from organoids
infected with SARS-CoV-2.
Does not address how cirrhotic liver or
alcohol/smoking/obesity-associated liver organoids respond to
SARS-CoV-2 infectivity and replication, worth pursuing to
experimentally address clinical data indicating co-morbidities.
14.64.4 Significance
Useful model to rapidly study drug activity against SARS-CoV-2
infection in liver, while monitoring baseline liver damage.
Liver abnormality observed in >50% of CoVID-19 patients; the
results from this study could explain the bile acid accumulation and
consequent liver damage observed.
14.64.5 Credit
Review by Samarth Hegde as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.65 The sequence of human ACE2 is suboptimal for binding the S spike protein of SARS coronavirus 2
Severe Acute Respiratory Syndrome Coronavirus 2
(SARS-CoV-2) infects cells through S spike glycoprotein binding
angiotensin-converting enzyme (ACE2) on host cells. S protein can bind
both membrane-bound ACE2 and soluble ACE2 (sACE2), which can serve as a
decoy that neutralizes infection. Recombinant sACE2 is now being tested
in clinical trials for COVID-19. To determine if a therapeutic sACE2
with higher affinity for S protein could be designed, authors generated
a library containing every amino acid substitution possible at the 117
sites spanning the binding interface with S protein. The ACE2 library
was expressed in human Expi293F cells and cells were incubated with
medium containing the receptor binding domain (RBD) of SARS-CoV-2 fused
to GFP. Cells with high or low affinity mutant ACE2 receptor compared to
affinity of wild type ACE2 for the RBD were FACS sorted and transcripts
from these sorted populations were deep sequenced. Deep mutagenesis
identified numerous mutations in ACE2 that enhance RBD binding. This
work serves to identify putative high affinity ACE2 therapeutics for the
treatment of CoV-2.
14.65.3 Limitations
The authors generated a large library of mutated
ACE2, expressed them in human Expi293F cells, and performed deep
mutagenesis to identify enhanced binders for the RBD of SARS-CoV-2 S
protein. While these data serve as a useful resource, the ability of the
high affinity ACE2 mutants identified to serve as therapeutics needs
further validation in terms of conformational stability when purified as
well as efficacy/safety both in vitro and in vivo. Additionally,
authors mentioned fusing the therapeutic ACE2 to Fc receptors to elicit
beneficial host immune responses, which would need further design and
validation.
14.65.4 Significance
This study identified structural ACE2 mutants that
have potential to serve as therapeutics in the treatment of SARS-CoV-2
upon further testing and validation.
14.65.5 Credit
This review was undertaken by Katherine Lindblad and Tamar Plitt as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
Title: A serological assay to detect SARS-Cov-2 seroconversion in
humans
Note: the authors of this review work in the same institution as the
authors of the study
Main findings:
Production of recombinant whole Spike (S) protein and the smaller
Receptor Binding Domain (RBD) based on the sequence of Wuhan-Hu-1
SARS-CoV-2 isolate. The S protein was modified to allow trimerization
and increase stability. The authors compared the antibody reactivity of
59 banked human serum samples (non-exposed) and 3 serum samples from
confirmed SARS-CoV-2 infected patients. All Covid-19 patient sera
reacted to the S protein and RBD domain compared to the control sera.
The authors also characterized the antibody isotypes from the Covid-19
patients, and observed stronger IgG3 response than IgG1. IgM and IgA
responses were also prevalent.
Limitations of the study:
The authors analyzed a total of 59 control human serum samples, and
samples from only three different patients to test for reactivity
against the RBD domain and full-length spike protein. It will be
important to follow up with a larger number of patient samples to
confirm the data obtained. Furthermore, it would be interesting to
assess people at different age groups and determine whether unexposed
control kids have a higher “background”.
Applications of the assay described in this study in diagnosis are
limited, since antibody response should start to be detectable only one
to two weeks after infection. Future studies will be required to assess
how long after infection this assay allow to detect anti-CoV2
antibodies. Finally, while likely, the association of seroconversion
with protective immunity against SARS-Cov-2 infection still needs to be
fully established.
Relevance:
This study has strong implications in the research against SARS-Cov-2.
First, it is now possible to perform serosurveys and determine who has
been infected, allowing a more accurate estimate of infection prevalence
and death rate. Second, if it is confirmed that re-infection does not
happen (or is rare), this assay can be used as a tool to screen
healthcare workers and prioritize immune ones to work with infected
patients. Third, potential convalescent plasma donors can now be
screened to help treating currently infected patients. Of note, this
assay does not involve live virus handling. experimentally, this is an
advantage as the assay does not require the precautions required by
manipulation of live virus. Finally, the recombinant proteins described
in this study represent new tools that can be used for further
applications, including vaccine development.
14.66 COMPARATIVE PATHOGENESIS OF COVID-19, MERS AND SARS IN A NON-HUMAN PRIMATE MODEL
This work assesses SARS-CoV-2 infection in young adult and aged
cynomolgus macaques. 4 macaques per age group were infected with
low-passage clinical sample of SARS-CoV-2 by intranasal and
intratracheal administration. Viral presence was assessed in nose,
throat and rectum through RT-PCR and viral culture. SARS-CoV-2
replication was confirmed in the respiratory track (including nasal
samples), and it was also detected in ileum. Viral nucleocapsid
detection by IHC showed infection of type I and II pneumocytes and
epithelia. Virus was found to peak between 2 and 4 days after
administration and reached higher levels in aged vs. young animals. The
early peak is consistent with data in patients and contrasts to SARS-CoV
replication. SARS-CoV-2 reached levels below detection between 8 and 21
days after inoculation and macaques established antibody immunity
against the virus by day 14. There were histopathological alteration in
lung, but no overt clinical signs. At day 4 post inoculation of
SARS-CoV-2, two of four animals presented foci of pulmonary
consolidation, with limited areas of alveolar edema and pneumonia, as
well as immune cell infiltration. In sum, cynomolgus macaques are
permissive to SARS- CoV-2 and develop lung pathology (less severe than
SARC-CoV, but more severe than MERS-CoV).
14.66.3 Limitations
Even though cynomolgus macaques were permissive to SARS-CoV-2
replication, it is unclear if the viral load reaches levels comparable
to humans and there wasn’t overt clinical pathology.
14.66.4 Significance
The development of platforms in which to carry out relevant
experimentation on SARS-CoV-2 pathophysiology is of great urgency.
Cynomolgus macaques offer an environment in which viral replication can
happen, albeit in a limited way and without translating into clinically
relevant symptoms. Other groups are contributing to SARS-CoV2 literature
using this animal model (2229), potentially showing protection against
reinfection in cured macaques. Therefore, this platform could be used to
examine SARS-CoV2 pathophysiology while studies in other animal models
are also underway (2168, 2244).
14.66.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.67 Investigating the Impact of Asymptomatic Carriers on COVID-19 Transmission
Multiple studies reported the same level of infectiousness
between symptomatic and asymptomatic carriers of SARS-CoV-2. Given that
asymptomatic and undocumented carriers escape public health surveillance
systems, a better mathematical model of transmission is needed to
determine a more accurate estimate of the basic reproductive number
(R0) of the virus to assess the contagiousness of virus. The authors
developed a SEYAR dynamical model for transmission of the new
coronavirus that takes into account asymptomatic and undocumented
carriers. The model was validated using data reported from thirteen
countries during the first three weeks of community transmission. While
current studies estimate R0 to be around 3, this model indicates that
the value could range between 5.5 to 25.4.
14.67.3 Limitations
The SEYAR model realistically depicts
transmission of the virus only during the initial stages of the disease.
More data is necessary to better fit the model with current trends. In
addition, multiple factors (e.g. behavioral patterns, surveillance
capabilities, environmental and socioeconomic factors) affect
transmission of the virus and so, these factors must be taken into
consideration when estimating the R0.
14.67.4 Significance
Public health authorities use the basic reproductive
number to determine the severity of disease. An accurate estimate of
R0 will inform intervention strategies. This model can be applied to
different locations to assess the potential impact of COVID-19.
14.67.5 Credit
This review was undertaken by Tamar Plitt and Katherine Lindblad as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.68 Antibody responses to SARS-CoV-2 in COVID-19 patients: the perspective application of serological tests in clinical practice
This study investigated the profile of the acute antibody response
against SARS-CoV-2 and provided proposals for serologic tests in
clinical practice. Magnetic Chemiluminescence Enzyme Immunoassay was
used to evaluate IgM and IgG seroconversion in 285 hospital admitted
patients who tested positive for SARS-CoV-2 by RT-PCR and in 52 COVID-19
suspected patients that tested negative by RT-PCR. A follow up study
with 63 patients was performed to investigate longitudinal effects. In
addition, IgG and IgM titers were evaluated in a cohort of close
contacts (164 persons) of an infected couple.
The median day of seroconversion for both IgG and IgM was 13 days after
symptom onset. Patients varied in the order of IgM/ IgG seroconversion
and there was no apparent correlation of order with age, severity, or
hospitalization time. This led the authors to conclude that for
diagnosis IgM and IgG should be detected simultaneously at the early
phase of infection.
IgG titers, but not IgM titers were higher in severe patients compared
to non-severe patients after controlling for days post-symptom onset.
Importantly, 12% of COVID-19 patients (RT-PCR confirmed) did not meet
the WHO serological diagnosis criterion of either seroconversion or
> 4-fold increase in IgG titer in sequential samples. This suggests
the current serological criteria may be too stringent for COVID-19
diagnosis.
Of note, 4 patients from a group of 52 suspects (negative RT-PCR test)
had anti-SARS-Cov-2 IgM and IgG. Similarly, 4.3% (7/162) of “close
contacts” who had negative RT-PCR tests were positive for IgG and/or
IgM. This highlights the usefulness of a serological assay to identify
asymptomatic infections and/or infections that are missed by RT-PCR.
14.68.3 Limitations
This group’s report generally confirms the findings of others that have
evaluated the acute antibody response to SARS-Cov-2. However, these data
would benefit from inclusion of data on whether the participants had a
documented history of viral infection. Moreover, serum samples that were
collected prior to SARS-Cov-2 outbreak from patients with other viral
infections would serve as a useful negative control for their assay.
Methodological limitations include that only one serum sample per case
was tested as well as the heat inactivation of serum samples prior to
testing. It has previously been reported that heat inactivation
interferes with the level of antibodies to SARS-Cov-2 and their protocol
may have resulted in diminished quantification of IgM, specifically (2247).
14.68.4 Significance
Understanding the features of the antibody responses against SARS-CoV is
useful in the development of a serological test for the diagnosis of
COVID-19. This paper addresses the need for additional screening methods
that can detect the presence of infection despite lower viral titers.
Detecting the production of antibodies, especially IgM, which are
produced rapidly after infection can be combined with PCR to enhance
detection sensitivity and accuracy and map the full spread of infection
in communities, Moreover, serologic assays would be useful to screen
health care workers in order to identify those with immunity to care for
patients with COVID19.
14.68.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.69 SARS-CoV-2 specific antibody responses in COVID-19 patients
Antibodies specific to SARS-CoV-2 S protein, the S1 subunit and the RBD (receptor-binding domain) were detected in all SARS-CoV-2 patient sera by 13 to 21 days post onset of disease. Antibodies specific to SARS-CoV N protein (90% similarity to SARS-CoV-2) were able to neutralize SARS-CoV-2 by PRNT (plaque reduction neutralizing test). SARS-CoV-2 serum cross-reacted with SARS-CoV S and S1 proteins, and to a lower extent with MERS-CoV S protein, but not with the MERS-CoV S1 protein, consistent with an analysis of genetic similarity. No reactivity to SARS-CoV-2 antigens was observed in serum from patients with ubiquitous human CoV infections (common cold) or to non-CoV viral respiratory infections.
14.69.3 Limitations
Authors describe development of a serological ELISA based assay for the detection of neutralizing antibodies towards regions of the spike and nucleocapsid domains of the SARS-CoV-2 virus. Serum samples were obtained from PCR-confirmed COVID-19 patients. Negative control samples include a cohort of patients with confirmed recent exposure to non-CoV infections (i.e. adenovirus, bocavirus, enterovirus, influenza, RSV, CMV, EBV) as well as a cohort of patients with confirmed infections with ubiquitous human CoV infections known to cause the common cold. The study also included serum from patients with previous MERS-CoV and SARS-CoV zoonotic infections. This impressive patient cohort allowed the authors to determine the sensitivity and specificity of the development of their in-house ELISA assay. Of note, seroconversion was observed as early as 13 days following COVID-19 onset but the authors were not clear how disease onset was determined.
14.69.4 Significance
Validated serological tests are urgently needed to map the full spread of SARS-CoV-2 in the population and to determine the kinetics of the antibody response to SARS-CoV-2. Furthermore, clinical trials are ongoing using plasma from patients who have recovered from SARS-CoV-2 as a therapeutic option. An assay such as the one described in this study could be used to screen for strong antibody responses in recovered patients. Furthermore, the assay could be used to screen health care workers for antibody responses to SARS-CoV-2 as personal protective equipment continues to dwindle. The challenge going forward will be to standardize and scale-up the various in-house ELISA’s being developed in independent laboratories across the world.
14.70 A brief review of antiviral drugs evaluated in registered clinical trials for COVID-19
Summary of clinical trials registered as
of March7, 2020 from U.S, Chinese, Korean, Iranian and European
registries. Out of the 353 studies identified, 115 were selected for
data extraction. 80% of the trials were randomized with parallel
assignment and the median number of planned inclusions was 63 (IRQ,
36-120). Most frequent therapies in the trials included; 1) antiviral
drugs [lopinavir/ritonavir (n-15); umifenovir (n=9); favipiravir (n=7);
redmesivir (n=5)]; 2) anti-malaria drugs [chloroquine (n-11);
hydroxychloroquine (n=7)}; immunosuppressant drugs [methylprednisolone
(n=5)]; and stem cell therapies (n=23). Medians of the total number of
planned inclusions per trial for these therapies were also included.
Stem cells and lopunavir/ritonavir were the most frequently evaluated
candidate therapies (23 and 15 trials respectively), whereas remdesivir
was only tested in 5 trials but these trials had the highest median
number of planned inclusions per trial (400, IQR 394-453). Most of the
agents used in the different trials were chosen based on preclinical
assessments of antiviral activity against SARS CoV and MERS Cov corona
viruses.
The primary outcomes of the studies were clinical (66%); virological
(23%); radiological (8%); or immunological (3%). The trials were
classified as those that included patients with severe disease only;
trials that included patients with moderate disease; and trials that
included patients with severe or moderate disease.
14.70.3 Limitations
The trials evaluated provided incomplete information:
23% of these were phase IV trials but the bulk of the trials (54%) did
not describe the phase of the study. Only 52% of the trials (n=60)
reported treatment dose and only 34% (n=39) reported the duration. A lot
of the trials included a small number of patients and the trials are
still ongoing, therefore no insight was provided on the outcome of the
trials.
14.70.4 Significance
Nonetheless, this review serves as framework for
identifying COVID-19 related trials, which can be expanded upon as new
trials begin at an accelerated rate as the disease spreads around the
world.
14.70.5 Credit
This review was undertaken by K Alexandropoulos as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.71 ACE-2 Expression in the Small Airway Epithelia of Smokers and COPD Patients: Implications for COVID-19
In bronchial epithelial samples from 3 different cohorts of
individuals, ACE-2 gene expression was found to be significantly
increased in both COPD patients and smokers relative to healthy
controls. Across all test subjects, ACE-2 gene expression was also
highly correlated with decreased forced expiratory volume in 1 second
(FEV1), which may explain the increased COVID-19 disease severity in
COPD patients. Former smokers were also found to show decreased ACE2
expression relative to current smokers and had no significant difference
when compared to non-smokers.
14.71.3 Limitations
While the upregulation of ACE-2 is an interesting
hypothesis for COVID-19 disease severity in COPD patients, this study
leaves many more unanswered questions than it addresses. Further studies
are required to show whether the specific cell type isolated in these
studies is relevant to the pathophysiology of COVID-19. Furthermore,
there is no attempt to show whether that increased ACE-2 expression
contributes to greater disease severity. Does the increased ACE-2
expression lead to greater infectivity with SARS-CoV-2? There is no
mechanistic explanation for why ACE-2 levels are increased in COPD
patients. The authors could also have considered the impact of
co-morbidities and interventions such as corticosteroids or
bronchodilators on ACE-2 expression. Finally, given the extensive
sequencing performed, the authors could have conducted significantly
more in-depth analyses into gene signature differences.
14.71.4 Significance
This study attempts to address an
important clinical finding that both smokers and COPD patients show
increased mortality from COVID-19. The novel finding that ACE-2
expression is induced in smokers and COPD patients suggests not only a
mechanism for the clinical observation, but also highlights the
potential benefit of smoking cessation in reducing the risk of severe
COVID-19 disease.
14.71.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.72 Dynamic profile of severe or critical COVID-19 cases
Authors evaluate clinical correlates of 10 patients (6 male
and 4 female) hospitalized for severe Acute Respiratory Syndrome
Coronavirus 2 (SARS-CoV-2). All patients required oxygen support and
received broad spectrum antibiotics and 6 patients received anti-viral
drugs. Additionally, 40% of patients were co-infected with influenza A.
All 10 patients developed lymphopenia, two of which developed
progressive lymphopenia (PLD) and died. Peripheral blood (PB)
lymphocytes were analyzed – low CD4 and CD8 counts were noted in most
patients, though CD4:CD8 ratio remained normal.
14.72.3 Limitations
The authors evaluated a small cohort of severe
SARS-CoV-2 cases and found an association between T cell lymphopenia and
adverse outcomes. However, this is an extremely small and diverse cohort
(40% of patients were co-infected with influenza A). These findings need
to be validated in a larger cohort. Additionally, the value of this data
would be greatly increased by adding individual data points for each
patient as well as by adding error bars to each of the figures.
14.72.4 Significance
This study provides a collection of clinical data and
tracks evolution of T lymphocyte in 10 patients hospitalized for
SARS-CoV-2, of which 4 patients were co-infected with influenza A.
14.72.5 Credit
This review was undertaken by Katherine Lindblad and Tamar Plitt as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.73 Association between Clinical, Laboratory and CT Characteristics and RT-PCR Results in the Follow-up of COVID-19 patients
Data analyzed from 52 COVID-19 patients admitted
and then discharged with COVID-19. Clinical, laboratory, and
radiological data were longitudinally recorded with illness timecourse
(PCR + to PCR-) and 7 patients (13.5%) were readmitted with a follow up
positive test (PCR+) within two weeks of discharge.
14.73.3 Main Findings
At admission:
The majority of patients had increased CRP at admission (63.5%).
LDH, and HSST TNT were significantly increased at admission.
Radiographic signs via chest CT showed increased involvement in
lower lobes: right lower lobe (47 cases, 90.4%), left lower lobe
(37 cases, 71.2%).
GGO (90.4%), interlobular septal thickening (42.3%), vascular
enlargement (42.3%), and reticulation (11.5%) were most commonly
observed.
After negative PCR test (discharge):
CRP levels decreased lymphocyte counts (#/L) increased
significantly (CD3+, CD3+/8+ and CD3+/4+) after negative PCR.
Consolidation and mixed GGO observed in longitudinal CT imaging
w different extents of inflammatory exudation in lungs, with
overall tendency for improvement (except 2/7 patients that were
readmitted after discharge with re-positive test) after negative
PCR.
Seven patients repeated positive RT-PCR test and were readmitted to
the hospital (9 to 17 day after initial discharge).
Follow up CT necessary to monitor improvement during recovery
and patients with lesion progression should be given more
attention.
Dynamic CT in addition to negative test essential in clinical
diagnosis due to nasal swab PCR sampling bias (false-negatives).
Increase in CRP occurred in 2 readmitted patients (and decr. in
lymphocytes in one patient), but was not correlated with new
lesions or disease progression vs. improvement (very low N).
Patients readmitted attributed to false-negative PCR vs.
re-exposure.
14.73.4 Limitations
Patients sampled in this study were generally
younger (65.4% < 50 yrs) and less critically ill/all discharged.
Small number of recovered patients (N=18). Time of follow up was
relatively short.. Limited clinical information available about patients
with re-positive test (except CRP and lymph tracking).
14.73.5 Extended Results
NOTE: Patients sampled in this study were generally younger (65.4%
< 50 yrs) and less critically ill/all discharged. After two
consecutive negative PCR tests, patients were discharged.
Clinical Results at Admission
Median interval disease onset to admission (5 days, IQR: 3-7)
Most common symptoms included fever, fatigue, dry cough, and
expectoration.
Fifteen patients had reduced lymphocyte counts (28.8%).
No change in WBC or Neutrophil counts.
The majority of patients had increased CRP at admission (63.5%).
LDH, and HSST TNT were significantly increased at admission.
Fibrinogen was trending high though not significant.
No major changes in liver function observed.
Radiographic signs via chest CT showed increased involvement in
lower lobes: right lower lobe (47 cases, 90.4%), left lower lobe (37
cases, 71.2%).
GGO (90.4%), interlobular septal thickening (42.3%), vascular
enlargement (42.3%), and reticulation (11.5%) were most commonly
observed.
Change in Clinical Results following Negative Test
CRP levels decreased after negative PCR.
Lymphocyte counts (#/L) increased significantly (CD3+, CD3+/8+
and CD3+/4+).
No significant change to CD4/8 ratio.
LDH, HSST TNT, and Fibronegin remained high throughout, though range
observed decreased over time.
Consolidation and mixed GGO observed in longitudinal CT imaging.
Patients showed different extents of inflammatory exudation in
lungs, with overall tendency for improvement (except 2/7 patients
that were readmitted after discharge with re-positive test).
Patients Readmitted with PCR+ test
Seven patients repeated positive RT-PCR test and were readmitted
to the hospital (9 to 17 day after initial discharge).
Improvement during readmission in 4 patients and observation of
segmental progression CT in 2 patients (2/18 or 11.1% - re-positive
9 and 10 days post-discharge).
Two patients showed new GGO, while others improved greatly.
Follow up CT necessary to monitor improvement during recovery and
patients with lesion progression should be given more attention.
Dynamic CT in addition to negative test essential in clinical
diagnosis due to nasal swab PCR sampling bias (false-negatives).
Increase in CRP occurred in 2 readmitted patients (and decr. in
lymphocytes in one patient), but was not correlated with new lesions
or disease progression vs. improvement (very low N).
14.73.6 Significance
Study tracked key clinical features associated with
disease progression, recovery, and determinants of clinical
diagnosis/management of COVID-19 patients.
14.73.7 Credit
This review was undertaken by Natalie Vaninov as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.74 An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 and multiple 2 endemic, epidemic and bat coronavirus
ribonucleoside analog β-D-N4 30 hydroxycytidine (NHC)
Remdesivir
14.74.2 Main Findings
β-D-N4 30 –hydroxycytidine (NHC, EIDD-1931) is an
orally bioavailable ribonucleoside with antiviral activity against
various RNA viruses including Ebola, Influenza and CoV. NHC activity
introduceds mutations in the viral (but not cellular) RNA in a
dose dependent manner that directly correlated with a decrease in
viral titers. Authors show that NHC inhibited multiple genetically
distinct Bat-CoV viruses in human primary epithelial cells without
affectingcell viability even at high concentrations (100µM).
Prophylactic oral administration of NHC in C57BL/6 mice reduce lung
titers of SARS-CoV and prevented weight loss and hemorrhage. Therapeutic
administration of NHC in C57BL/6 mice 12 hours post infected with
SARS-CoV reduced acute lung injury, viral titer, and lung hemorrhage.
The degree of clinical benefit was dependent on the time of treatment
initiation post infection. The authors also demonstrate that NHC reduces
MERS-CoV infection titers, pathogenesis, and viral RNA in prophylactic
and therapeutic settings.
14.74.3 Limitations
Most of the experiments were conducted using MERS-CoV, and
SARS-CoV and a few experiments were conducted using other strains of CoV
as opposed to SARS-CoV-2. The authors note the core residues that make
up the RNA interaction sites (which constitutes the NHC interaction
sites) are highly conserved among CoV and because of this conservation
their understanding is that NHC can inhibit a broad-spectrum of CoV
including SARS-CoV-2.
The increased viral mutation rates associated with NHC activity may have
adverse effects if mutations cause the virus to become drug resistant,
more infectious or speed-up immune evasion. In addition, the temporal
diminishing effectiveness of NHC on clinical outcome when NHC was used
therapeutically is concerning. However, the longer window (7-10 days)
for clinical disease onset in human patients from the time of infection
compared to that of mice (24-48 hours), may associate with increased NHC
effectiveness in the clinic.
14.74.4 Significance
Prophylactic or therapeutic oral administration of NHC
reduces lung titers and prevents acute lung failure in C57B\6 mice
infected with CoV. Given its broad-spectrum antiviral activity, NHC
could turn out to be a useful drug for treating current, emerging and
future corona virus outbreaks.
#### Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.75 Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs
A panel of ~3,000 FDA- and IND-approved
antiviral drugs were previously screened for inhibitory efficacy against
SARS CoV, a coronavirus related to the novel coronavirus SARS CoV-2
(79.5%) homology. 35 of these drugs along with another 15 (suggested by
infectious disease specialists) were tested in vitro for their ability
to inhibit SARS CoV-2 infectivity of Vero cells while preserving cell
viability. The infected cells were scored by immunofluorescence analysis
using an antibody against the N protein of SARS CoV-2. Chloroquine,
lopinavir and remdesivir were used as reference drugs.
Twenty four out of 50 drugs exhibited antiviral activity with IC50
values ranging from 0.1-10µM. Among these, two stood ou: 1) the-anti
helminthic drug niclosamide which exhibited potent antiviral activity
against SARS CoV-2 (IC50=0.28 µM). The broad-spectrum antiviral effect
of niclosamide against SARS and MERS-CoV have been previously documented
and recent evidence suggests that in may inhibit autophagy and reduce
MERS C0V replication. 2) Ciclesonide, a corticosteroid used to treat
asthma and allergic rhinitis, also exhibited antiviral efficacy but with
a lower IC50 (4.33µM) compared to niclosamide. The antiviral effects of
ciclesonide were directed against NSP15, a viral riboendonuclease which
is the molecular target of this drug.
14.75.3 Limitations
The drugs were tested against SARS CoV-2 infectivity in
vitro only, therefore preclinical studies in animals and clinical trials
in patients will be need for validation of these drugs as therapeutic
agents for COVID-19. In addition, niclosamide exhibits low adsorption
pharmatokinetically which could be alleviated with further development
of drug formulation to increase effective delivery of this drug to
target tissues. Nonetheless, niclosamide and ciclesonide represent
promising therapeutic agents against SARS CoV-2 given that other
compounds tested in the same study including favipiravir (currently used
in clinical trials) and atazanavir (predicted as the most potent
antiviral drug by AI-inference modeling) did not exhibit antiviral
activity in the current study.
14.75.4 Credit
This review was undertaken by K Alexandropoulos as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.76 Respiratory disease and virus shedding in rhesus macaques inoculated with SARS-CoV-2
animal model, pulmonary infiltrates, dynamic
of antibody response, cytokine
14.76.2 Main Findings
Inoculation of 8 Resus macaques with SARS-Cov-2 , which
all showed clinical signs of infection (respiratory pattern, reduced
appetite, weight loss, elevated body temperature) resulting in moderate,
transient disease. Four animals were euthanized at 3dpi, the 4 others at
21 dpi. Study of viral loads in different organs showed that nose swab
and throat swabs were the most sensitive, with bronchio-alveolar lavage.
Interstitial pneumonia was visible in radiographies and at the
histological scale too. Clinically, the macaques had similar symptoms as
described in human patients with moderate disease.
Viral shedding was consistently detected in nose swabs and throat swabs
immediately after infection but less consistent thereafter which could
reflect virus administration route (intranasal, oral). Bronchoalveolar
lavages performed as a measure of virus replication in the lower
respiratory tract on animals maintained for 21 days, contained high
viral loads in 1 and 3dpi. The majority of the animals exhibited
pulmonary edema and mild to moderate interstitial pneumonia on terminal
bronchioles. In addition to the lung, viral RNA could also be detected
throughout the respiratory track where viral replication mainly
occurred.
Immunologic responses included leukocytosis, neutrophilia, monocytosis
and lymphopenia in the majority of the animals at 1dpi. Lymphocytes and
monocytes re-normalized at 2dpi. Neutrophils declined after 3dpi and
through 10dpi after which they started to recover. After infection,
serum analysis revealed significant increases in IL1ra, IL6, IL10,
IL15, MCP-1, MIP-1b, but quick normalization (3dpi). Antibody
response started around 7dpi, and the antibody titers stayed elevated
until 21dpi (day of animal euthanasia).
14.76.3 Limitations
The macaques were inoculated via a combination
of intratracheal, intranasal, ocular and oral routes, which might not
reproduce how humans get infected. Maybe this can lead to different
dynamics in the host immune response. Also, the authors noted that the
seroconversion was not directly followed by a decline in viral loads, as
observed in covid19 patients.
14.76.4 Significance
This work confirms that rhesus
macaques can be a good model to study Covid-19, as it has been shown by
other groups (2229, 2243, 2256). While these experiments recapitulate
moderate COVID-19 in humans, the mode of inoculation via a combination
of intratracheal, intranasal, ocular and oral routes, might not
reproduce how humans get infected and may lead to different dynamics in
the host immune response. For example, the authors noted that the
seroconversion was not directly followed by a decline in viral loads, as
observed in COVID-19 patients. Therefore, it will be interesting to
follow their antibody titers longer and further assess the
possibility/effect of reinfection in these macaques. It is essential to
be able to understand the dynamic of the disease and associated immune
responses, and to work on vaccine development and antiviral drug
testing.
14.76.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.77 ACE2 Expression is Increased in the Lungs of Patients with Comorbidities Associated with Severe COVID-19
Transcriptomic analysis using systems-level meta-analysis and
network analysis of existing literature to determine ACE2 regulation
in patients who have frequent COVID-19 comorbidities [eg-
cardiovascular diseases, familial pulmonary hypertension, cancer].
Enrichment analyses indicated pathways associated with inflammation,
metabolism, macrophage autophagy, and ER stress.
ACE2 higher in adenocarcinoma compared to adjacent normal lung; ACE2
higher in COPD patients compared to normal.
Co-expression analysis identified genes important to viral entry
such as RAB1A, ADAM10, HMGBs, and TLR3 to be associated with ACE2 in
diseased lungs.
ACE2 expression could be potentially regulated by enzymes that
modify histones, including HAT1, HDAC2, and KDM5B.
14.77.3 Limitations:
Not actual CoVID-19 patients with co-morbidities, so interpretations
in this study need to be confirmed by analyzing upcoming
transcriptomics from CoVID-19 patients having co-morbidity metadata.
As mentioned by authors, study does not look at diabetes and
autoimmunity as risk factors in CoVID-19 patients due to lack of
data; would be useful to extend such analyses to those datasets when
available.
Co-expression analysis is perfunctory and needs
validation-experiments especially in CoVID-19 lung samples to mean
anything.
Epigenomic analyses are intriguing but incomplete, as existence of
histone marks does not necessarily mean occupancy. Would be
pertinent to check cell-line data (CCLE) or actual CoVID-19 patient
samples to confirm ACE2 epigenetic control.
14.77.4 Significance
Study implies vulnerable populations have ACE2 upregulation that
could promote CoVID-19 severity. Shows important data-mining
strategy to find gene-networks associated with ACE2 upregulation in
co-morbid patients.
Several of the genes co-upregulated with ACE2 in diseased lung might
play an important role in CoVID-19 and can be preliminary targets
for therapeutics
If in silico findings hold true, epigenetic control of ACE2
expression could be a new target for CoVID-19 therapy with
strategies such as KDM5 demethylases.
14.77.5 Credit
Review by Samarth Hegde as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
This work is based on previous work by the same group
that demonstrated that SARS-CoV-2can also enter host cells via CD147
(also called Basigin, part of the immunoglobulin superfamily, is
expressed by many cell types) consistent with their previous work with
SARS-CoV-1. 1 A prospective clinical trial was conducted with 17
patients receiving Meplazumab, a humanized anti-CD147 antibody, in
addition to all other treatments. 11 patients were included as a control
group (non-randomized).
They observed a faster overall improvement rate in the Meplazumab group
(e.g. at day 14 47% vs 17% improvement rate) compared to the control
patients. Also, virological clearance was more rapid with median of 3
days in the Meplazumab group vs 13 days in control group. In laboratory
values, a faster normalization of lymphocyte counts in the Meplazumab
group was observed, but no clear difference was observed for CRP levels.
14.78.3 Limitations
While the results from the study are encouraging, this
study was non-randomized, open-label and on a small number of patients,
all from the same hospital. It offers evidence to perform a larger scale
study. Selection bias as well as differences between treatment groups
(e.g. age 51yo vs 64yo) may have contributed to results. The authors
mention that there was no toxic effect to Meplazumab injection but more
patient and longer-term studies are necessary to assess this.
14.78.4 Significance
These results seem promising as for now there are
limited treatments for Covid-19 patients, but a larger cohort of patient
is needed. CD147 has already been described to facilitate HIV (2259),
measles virus (2260), and malaria (2261) entry into host cells. This group was
the first to describe the CD147-spike route of SARS-Cov-2 entry in host
cells (2232) p147. Indeed, they had previously shown in 2005 that SARS-Cov
could enter host cells via this transmembrane protein (2262). Further
biological understanding of how SARS-CoV-2 can enter host cells and how
this integrates with ACE2R route of entry is needed. Also, the specific
cellular targets of the anti-CD147 antibody need to be assessed, as this
protein can be expressed by many cell types and has been shown to
involved in leukocytes aggregation (2263). Lastly, Meplazumab is not a
commercially-available drug and requires significant health resources to
generate and administer which might prevent rapid development and use.
14.78.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.79 Potent human neutralizing antibodies elicited 1 by SARS-CoV-2 infection
In this study the authors report the affinity, cross reactivity (with
SARS-CoV and MERS-CoV virus) and viral neutralization capacity of 206
monoclonal antibodies engineered from isolated IgG memory B cells of
patients suffering from SARS-CoV-2 infection in Wuhan, China. All
patients but one recovered from disease. Interestingly, the patient that
did not recover had less SARS-CoV-2 specific B cells circulating
compared to other patients.
Plasma from all patients reacted to trimeric Spike proteins from
SARS-CoV-2, SARS-CoV and MERS-CoV but no HIV BG505 trimer. Furthermore,
plasma from patients recognized the receptor binding domain (RBD) from
SARS-CoV-2 but had little to no cross-reactivity against the RBD of
related viruses SARS-CoV and MERS-CoV, suggesting significant
differences between the RBDs of the different viruses. Negligible levels
of cross-neutralization using pseudoviruses bearing Spike proteins of
SARS-CoV-2, SARS-CoV or MERS-CoV, were observed, corroborating the ELISA
cross-reactivity assays on the RBDs.
SARS-CoV-2 RBD specific B cells constituted 0.005-0.065% of the total B
cell population and 0.023-0.329% of the memory subpopulation. SARS-CoV
specific IgG memory B cells were single cell sorted to sequence the
antibody genes that were subsequently expressed as recombinant IgG1
antibodies. From this library, 206 antibodies with different binding
capacities were obtained. No discernible patterns of VH usage were found
in the 206 antibodies suggesting immunologically distinct responses to
the infection. Nevertheless, most high-binding antibodies were derived
by clonal expansion. Further analyses in one of the patient derived
clones, showed that the antibodies from three different timepoints did
not group together in phylogenetic analysis, suggesting selection during
early infection.
Using surface plasmon resonance (SPR) 13 antibodies were found to have
10-8 tp 10-9 dissociation constants (Kd). Of the 13 antibodies, two
showed 98-99% blocking of SARS-CoV-2 RBD-ACE2 receptor binding in
competition assays. Thus, low Kd values alone did not predict ACE2
competing capacities. Consistent with competition assays the two
antibodies that show high ACE2 blocking (P2C-2F6 and P2C-1F11) were the
most capable of neutralizing pseudoviruses bearing SARS-CoV-2 spike
protein (IC50 of 0.06 and 0.03 µg/mL, respectively). Finally, using
SPR the neutralizing antibodies were found to recognize both overlapping
and distinct epitopes of the RBD of SARS-CoV-2.
14.79.3 Limitations
Relatively low number of patients
No significant conclusion can be drawn about the possible
> correlation between humoral response and disease severity
In vitro Cytopathic Effect Assay (CPE) for neutralization activity
Huh7 cells were used in neutralization assays with
> pseudoviruses, and they may not entirely mimic what happens in
> the upper respiratory tract
CPE assay is not quantitative
Duplicated panel in Figure 4C reported (has been fixed in version 2)
14.79.4 Significance
This paper offers an explanation as to why
previously isolated antibodies against SARS-CoV do not effectively block
SARS-CoV-2. Also, it offers important insight into the development of
humoral responses at various time points during the first weeks of the
disease in small but clinically diverse group of patients. Furthermore,
it provides valuable information and well characterized antibody
candidates for the development of a recombinant antibody treatment for
SARS-CoV-2. Nevertheless, it also shows that plasmapheresis might have
variability in its effectiveness, depending on the donor’s antibody
repertoire at the time of donation.
14.79.5 Credit
Review by Jovani Catalan-Dibene as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.80 Characterisation of the transcriptome and proteome of SARS-CoV-2 using direct RNA sequencing and tandem mass spectrometry reveals evidence for a cell passage induced in-frame deletion in the spike glycoprotein that removes the furin-like cleavage site.
The authors performed long read RNA sequencing using an Oxford Nanopore
MinION as well as tandem mass spec (MS) on Vero cells (a cell line
derived from kidney cells of the African green monkey that is deficient
in interferon) infected with SARS-CoV-2.
The authors found that passage of the virus in Vero cells gave rise to a
spontaneous 9 amino acid deletion (679-NSPRRARSV-687 to I) in the spike
(S) protein. The deleted sequence overlaps a predicted furin cleavage
site at the S1 / S2 domain boundary that is present in SARS-CoV-2 but
not SARS-CoV or the closely related bat coronavirus RaTG13, which are
cleaved at S1 / S2 by other proteases (16). Furin cleavage sites at
similar positions in other viruses have been linked to increased
pathogenicity and greater cell tropism (2265). Loss of this site in
SARS-CoV-2 has also already been shown to increase viral entry into Vero
but not BHK cells (which are also interferon deficient) (25). The
authors therefore make an important contribution in demonstrating that
passage in Vero cells may lead to spontaneous loss of a key
pathogenicity-conferring element in SARS-CoV-2.
14.80.3 Limitations
As the authors note, a similar study posted earlier by Kim et al., which
also passaged SARS-CoV-2 in Vero cells, did not identify any loss in the
S protein furin cleavage site (2266). It therefore remains to be
determined how likely it is that this mutation spontaneously arises. A
quantitative investigation using multiple experimental replicas to
understand the spontaneous viral mutation rate at this site and
elsewhere would be informative. Also, the mechanistic basis for the
higher viral fitness conferred by loss of the furin cleavage site in
Vero cells – but, evidently, not in vivo in humans, as this site is
maintained in all currently sequenced circulating isolates - remains to
be understood.
Due to the high base-call error rate of MinION sequencing, the authors’
bioinformatic pipeline required aligning transcripts to a reference to
correct sequencing artifacts. This presumably made it difficult or
impossible to identify other kinds of mutations, such as single
nucleotide substitutions, which may occur even more frequently than the
deletions identified in this work. Pairing long read sequencing with
higher-accuracy short-read sequencing may be one approach to overcome
this issue.
14.80.4 Significance
As the authors suggest, animal studies using live virus challenge may
need to periodically verify the genomic integrity of the virus, or
potentially risk unknowingly using a likely less-pathogenic variant of
the virus.
More broadly, the results emphasize the complexity and plasticity of the
SARS-CoV-2 viral transcriptome and proteome. For example, the authors
found multiple versions of transcripts encoding the nucleocapsid (N)
protein, each with different small internal deletions, some of which
were verified for translation by MS. A number of peptides arising from
translation of unexpected rearrangements of transcripts were also
detected. Additionally, the authors identified phosphorylation of a
number of viral proteins (N, M, ORF 3a, nsp3, nsp9, nsp12 and S). For
any cases where these have functional consequences, targeting the
kinases responsible could be an avenue for drug development.
Understanding the functional consequences of the mutations, transcript
variations, and post translational modifications identified in this
study will be important future work.
14.80.5 Credit
This review was undertaken by Tim O’Donnell, Maria Kuksin as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.81 A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug- Repurposing
Gordon et al cloned, tagged and expressed 26 of the 29
SARS-CoV-2 proteins individually in HEK293T cells and used mass
spectrometry to identify protein-protein interactions. They identified
332 viral-host protein-protein interactions. Furthermore, they used
these interactions to identify 66 existing drugs known to target host
proteins or host pathways (eg SARS-CoV-2 N and Orf8 proteins interact
with proteins regulated by the mTOR pathway, so mTOR inhibitors
Silmitasertib and Rapamycin are possible drug candidates).
14.81.3 Limitations
The main limitation of the study stems from
the reductionist model: overexpression of plasmids encoding individual
viral proteins in HEK293T cells. This precludes any interactions between
the viral proteins, or the combined effects of multiple proteins on the
host, as they are expressed individually. Moreover, HEK293T cells come
from primary embryonic kidney and therefore might not reflect how
SARS-CoV-2 interacts with its primary target, the lung. However, the
authors found that the proteins found to interact with viral proteins in
their experiments are enriched in lung tissue compared to HEK293Ts.
14.81.4 Significance
The authors provide a “SARS-CoV-2 interaction map,”
which may provide potential hypotheses as to how the virus interacts
with the host. Further, they identified existing drugs that could
disrupt these host-viral interactions and curb SARS-CoV-2 infection.
Although these interactions have not been validated, this paper acts as
a valuable resource.
14.81.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.82 First Clinical Study Using HCV Protease Inhibitor Danoprevir to Treat Naïve and Experienced COVID-19 Patients
The authors treated 11 Covid-19 patients with Danoprevir, a
commercialized HCV protease inhibitor (2269)(p4), boosted by ritonavir (2270), a
CYP3A4 inhibitor (which enhances the plasma concentration and
bioavailabilty of Danoprevir). Two patients had never received
anti-viral therapy before (=naïve), whereas nine patients were on
Lopinavir/Ritonavir treatment before switching to Danoprevir/Ritonavir
(=experienced). The age ranged from 18 to 66yo.
Naïve patients that received Danoprevir/Ritonavir treatment had a
decreased hospitalization time. Patients treated with
Lopinavir/Ritonavir did not have a negative PCR test, while after
switching to Danoprevir/Ritonavir treatment, the first negative PCR test
occurred at a median of two days.
14.82.3 Limitations
The results of the study are very hard to interpret as there is no
control group not receiving Danoprevir/Ritonavir treatment. This was
especially true in naïve patients who seemed to have more mild symptoms
before the start of the study and were younger (18 and 44yo) compared to
the experienced patients (18 to 66yo). The possibility that the patients
would have recovered without Danoprevir/Ritonavir treatment cannot be
excluded.
14.82.4 Significance
The authors of the study treated patients with Danoprevir, with the
rational to that this is an approved and well tolerated drug for HCV
patients (2270), and that it could also target the protease from SARS-CoV-2
(essential for viral replication and transcription). Indeed, homology
modelling data indicated that HCV protease inhibitors have the highest
binding affinity to Sars-Cov2 protease among other approved drugs (2271).
While this study shows that the combination of Danoprevir and Ritonavir
might be beneficial for Covid-19 patients, additional clinical trials
with more patients and with better methodology (randomization and
control group) are needed to make further conclusions.
14.82.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.83 Efficacy of hydroxychloroquine in patients with COVID-19: results of a randomized clinical trial
This is a randomized clinical trial of hydroxychloroquine (HCQ) efficacy
in the treatment of COVID-19. From February 4 – February 28, 2020 142
COVID-19 positive patients were admitted to Renmin Hospital of Wuhan
University. 62 patients met inclusion criteria and were enrolled in a
double blind, randomized control trial, with 31 patients in each arm.
Inclusion criteria:
Age ≥ 18 years
Positive diagnosis COVID-19 by detection of SARS-CoV-2 by RT-PCR
Diagnosis of pneumonia on chest CT
Mild respiratory illness, defined by SaO2/SPO2 ratio > 93% or
PaO2/FIO2 ratio > 300 mmHg in hospital room conditions (Note:
relevant clinical references described below.)
Hypoxia is defined as an SpO2 of 85-94%; severe hypoxia <
85%.
The PaO2/FIO2 (ratio of arterial oxygen tension to fraction
of inspired oxygen) is used to classify the severity of acute
respiratory distress syndrome (ARDS). Mild ARDS has a
PaO2/FIO2 of 200-300 mmHg, moderate is 100-200, and severe
< 100.
Willing to receive a random assignment to any designated treatment
group; not participating in another study at the same time
Exclusion criteria:
Severe or critical respiratory illness (not explicitly defined,
presumed to be respiratory function worse than outlined in inclusion
criteria); or participation in trial does not meet patient’s maximum
benefit or safe follow up criteria
Retinopathy or other retinal diseases
Conduction block or other arrhythmias
Severe liver disease, defined by Child-Pugh score ≥ C or AST >
twice the upper limit
Pregnant or breastfeeding
Severe renal failure, defined by eGFR ≤ 30 mL/min/1.73m2, or on
dialysis
Potential transfer to another hospital within 72h of enrollment
Received any trial treatment for COVID-19 within 30 days before the
current study
All patients received the standard of care: oxygen therapy, antiviral
agents, antibacterial agents, and immunoglobulin, with or without
corticosteroids. Patients in the HCQ treatment group received additional
oral HCQ 400 mg/day, given as 200 mg 2x/day. HCQ was administered from
days 1-5 of the trial. The primary endpoint was 5 days post enrollment
or a severe adverse reaction to HCQ. The primary outcome evaluated was
time to clinical recovery (TTCR), defined as return to normal body
temperature and cough cessation for > 72h. Chest CT were imaged on
days 0 and 6 of the trial for both groups; body temperature and patient
reports of cough were collected 3x/day from day 0 – 6. The mean age and
sex distribution between the HCQ and control arms were comparable.
14.83.3 Main Findings
There were 2 patients showing mild secondary effects of HCQ treatment.
More importantly, while 4 patients in the control group progressed to
severe disease, none progressed in the treatment group.
TTCR was significantly decreased in the HCQ treatment arm; recovery from
fever was shortened by one day (3.2 days control vs. 2.2 days HCQ, p =
0.0008); time to cessation of cough was similarly reduced (3.1 days
control vs. 2.0 days HCQ, p = 0.0016).
Overall, it appears that HCQ treatment of patients with mild COVID-19
has a modest effect on clinical recovery (symptom relief on average 1
day earlier) but may be more potent in reducing the progression from
mild to severe disease.
14.83.4 Limitations
This study is limited in its inclusion of only patients with mild
disease, and exclusion of those on any treatment other than the standard
of care. It would also have been important to include the laboratory
values of positive RT-PCR detection of SARS-CoV-2 to compare the
baseline and evolution of the patients’ viral load.
14.83.5 Limitations
Despite its limitations, the study design has good rigor as a double
blind RCT and consistent symptom checks on each day of the trail. Now
that the FDA has approved HCQ for treatment of COVID-19 in the USA, this
study supports the efficacy of HCQ use early in treatment of patients
showing mild symptoms, to improve time to clinical recovery, and
possibly reduce disease progression. However, most of the current
applications of HCQ have been in patients with severe disease and for
compassionate use, which are out of the scope of the findings presented
in this trial. Several additional clinical trials to examine
hydroxychloroquine
are now undergoing; their results will be critical to further validate
these findings.
14.83.6 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
Structure-based modeling of SARS-CoV-2 peptide/HLA-A02 antigens
The authors utilize homology modeling to identify peptides from the
SARS-CoV-2 proteome that potentially bind HLA-A*02:01.
They utilize high-resolution X-ray structures of peptide/MHC
complexes on Protein Data Bank, substitute homologous peptides with
SARS-CoV-2 peptides, and calculate MHC/SARS-CoV-2 peptide Rosetta
binding energy.
They select MHC/SARS-CoV-2 complex models with highest binding
energy for further study and publish models in an online database
(https://rosettamhc.chemistry.ucsc.edu).
Limitations:
The authors only utilize computational methods and predicted
SARS-CoV-2 peptides must be validated experimentally for
immunogenicity and clinical response.
Due to computational burden and limited availability of high
resolution X-ray structures on PDB, authors only simulate 9-mer and
10-mer peptide binding to HLA-A*02:01.
Since the authors compare select existing X-ray structures as a
starting point, backbone conformations that deviate significantly
between test and template peptides are not captured. Furthermore,
Rosetta modeling protocols do not capture all possible structures
and binding energy scoring does not fully recapitulate fundamental
forces.1,2
Importance/Relevance:
The authors identify and publish high-scoring SARS-CoV-2 peptides
that may direct a targeted, experimental validation approach toward
a COVID-19 vaccine.
The authors utilize Rosetta simulation to further filter results
from NetMHCpan 4.0, supporting machine learning prediction with
structural analysis.
The authors develop RosettaMHC, a computationally efficient method
of leveraging existing X-ray structures for identification of
immunogenic peptides.
References:
Bender, B. J., Cisneros, A., 3rd, Duran, A. M., Finn, J. A., Fu, D.,
Lokits, A. D., . . . Moretti, R. (2016). Protocols for Molecular
Modeling with Rosetta3 and RosettaScripts. Biochemistry, 55(34),
4748-4763. doi:10.1021/acs.biochem.6b00444
Alford, R. F., Leaver-Fay, A., Jeliazkov, J. R., O’Meara, M. J.,
DiMaio, F. P., Park, H., . . . Gray, J. J. (2017). The Rosetta
All-Atom Energy Function for Macromolecular Modeling and Design. J
Chem Theory Comput, 13(6), 3031-3048. doi:10.1021/acs.jctc.7b00125
Review by Jonathan Chung as part of a project by students, postdocs and
faculty at the Immunology Institute of the Icahn school of medicine,
Mount Sinai.
14.84 Serology characteristics of SARS-CoV-2 infection since the exposure and post symptoms onset
Currently, the diagnosis of SARS-CoV-2 infection
entirely depends on the detection of viral RNA using polymerase chain
reaction (PCR) assays. False negative results are common, particularly
when the samples are collected from upper respiratory. Serological
detection may be useful as an additional testing strategy. In this study
the authors reported that a typical acute antibody response was induced
during the SARS-CoV-2 infection, which was discuss earlier1. The
seroconversion rate for Ab, IgM and IgG in COVID-19 patients was 98.8%
(79/80), 93.8% (75/80) and 93.8% (75/80), respectively. The first
detectible serology marker was total antibody followed by IgM and IgG,
with a median seroconversion time of 15, 18 and 20 days-post exposure
(d.p.e) or 9, 10- and 12-days post-onset (d.p.o). Seroconversion was
first detected at day 7d.p.e in 98.9% of the patients. Interestingly
they found that viral load declined as antibody levels increased. This
was in contrast to a previous study (2212), showing that increased antibody
titers did not always correlate with RNA clearance (low number of
patient sample).
14.84.3 Limitations
Current knowledge of the antibody response to SAR-CoV-2
infection and its mechanism is not yet well elucidated. Similar to the
RNA test, the absence of antibody titers in the early stage of illness
could not exclude the possibility of infection. A diagnostic test, which
is the aim of the authors, would not be useful at the early time points
of infection but it could be used to screen asymptomatic patients or
patients with mild disease at later times after exposure.
14.84.4 Significance
Understanding the antibody responses against SARS-CoV2 is
useful in the development of a serological test for the diagnosis of
COVID-19. This manuscript discussed acute antibody responses which can
be deducted in plasma for diagnostic as well as prognostic purposes.
Thus, patient-derived plasma with known antibody titers may be used
therapeutically for treating COVID-19 patients with severe illness.
14.84.5 Credit
This review was undertaken and edited by Konstantina A as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.85 SARS-CoV-2 launches a unique transcriptional signature from in vitro, ex vivo, and in vivo systems
Given the high mortality rate of SARS-CoV-2 relative to
other respiratory viruses such seasonal IAV and RSV, there may be
underlying host-pathogen interactions specific to SARS-CoV-2 that
predispose to a worse clinical outcome. Using in vivo, ex vivo, and
in vitro systems, the authors profiled host cell transcriptional
responses to SARS-CoV-2 and to other common respiratory viruses
(seasonal IAV and RSV). SARS-CoV-2 infection in vitro led to an
induction of type I interferon response signaling and the upregulation
of cytokine/chemokines transcripts. In comparison with IAV and RSV
infection, SARS-CoV-2 in vitro appears to uniquely induce less type I
and type III interferon expression and higher levels of two cytokines
previously implicated in respiratory inflammation. Lastly, in vivo
data from ferrets showed a reduced induction of cytokines and chemokines
by SARS-CoV-2 infection relative to IAV infection.
14.85.3 Limitations
While these results are promising, there are several
key weaknesses of this paper. 1) As the authors point out, there is an
undetectable level of SARS-CoV-2 putative receptor (ACE2) and protease
(TMPRSS2) expression in the lung epithelial cell line used for the in
vitro studies. This raises the important question of whether viral
replication actually occurs in any of the models used, which may explain
the lack of interferon production observed in vitro in SARS-CoV-2
treated cells. Further studies characterizing viral titers across
timepoints are needed. 2) Furthermore, these studies only characterize
the host response at a single dose and timepoint per virus, and it is
unclear why these doses/timepoints were chosen. This leaves open the
possibility that the observed differences between viruses could be due
to differences in dose, timing, host response, or a combination of all
of these. 3) It is unclear whether ferrets are productively infected,
which cell types are infected, and the extent/timing of the clinical
course of infection. Moreover, the in vitro and in vivo data do not
strongly correlate and the reasons for this are unclear.
14.85.4 Significance
This paper describes potentially unique
transcriptional signatures of host cells exposed to SARS-CoV-2. If
validated, these findings may help explain clinical outcomes and could
be targeted in future therapeutic interventions.
14.85.5 Potential Conflicts of Interest Disclosure
The reviewers are also researchers at the Icahn School
of Medicine at Mount Sinai.
14.85.6 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.86 A New Predictor of Disease Severity in Patients with COVID-19 in Wuhan, China
377 hospitalized patients were divided into two groups: severe and
non-severe pneumonia. The laboratory results of their first day of
admission were retrospectively analyzed to identify predictors of
disease severity.
After adjusting for confounding factors from chronic comorbidities (such
as high blood pressure, type 2 diabetes, coronary heart disease, and
chronic obstructive pulmonary disease), the independent risk factors
identified for severe pneumonia were age, the ratio of
neutrophil/lymphocytes counts, CRP and D-dimer levels.
To further increase the specificity and sensibility of these markers,
they showed that their multiplication [(Neutrophil/lymphocyte count)
* CRP * D-dimer] was a better predictor of disease severity, with
higher sensitivity (95.7%) and specificity (63.3%), with a cutoff value
of 2.68.
14.86.3 Limitations
This study included 377 hospitalized patients. Among them, 45.6%
patients tested positive for SARS-Cov-2 nucleic acid test results, and
others were included in the study based on clinically diagnosis even if
the molecular diagnosis was negative. Thus, additional studies are
needed to verify this on a larger number of covid-19 certified patients
and the cutoff value might be adjusted. Also, all the patients that did
not have the clinical characteristics of severe pneumonia were included
in the non-severe pneumonia group, but usually patients are also divided
into moderate and mild disease.
Also, studying different subset of lymphocytes could lead to a more
specific predictor. Another study showed that the neutrophils to CD8+ T
cells ratio was a strong predictor of disease severity (2182). Another more
precise study showed that the percentage of helper T cells and
regulatory T cells decrease but the percentage of naïve helper T cells
increases in severe cases (2175). Taking these subpopulations into account
might make the predictor more powerful.
Other studies also noted an inverse correlation between disease severity
and LDH (2216) or IL6 (2225) levels, but the authors here do not discuss LDH nor
IL6 levels, although this could help to strengthen the predictor.
The study is based on the results obtained on the first day of
admission, studying the dynamic of the changes in patients might also be
interesting to better predict disease severity.
14.86.4 Significance
This study confirms that the neutrophil to lymphocyte ratio can be a
predictor of disease severity as shown by many others (2174, 2175, 2188). The
novelty here is that they show that a combination with other markers can
enhance the specificity and sensibility of the predictor, although the
study could be improved by taking into account sub-populations of
lymphocytes and more biological factors from patients such as LDH and
IL6.
14.86.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.87 Metabolic disturbances and inflammatory dysfunction predict severity of coronavirus disease 2019 (COVID-19): a retrospective study
Retrospective Study on 97 COVID-19 hospitalized patients
(25 severe and 72 non-severe) analyzing clinical and laboratory
parameter to predict transition from mild to severe disease based on
more accessible indicators (such as fasting blood glucose, serum protein
or blood lipid) than inflammatory indicators. In accordance with other
studies, age and hypertension were risk factors for disease severity,
and lymphopenia and increased IL-6 was observed in severe patients. The
authors show that fasting blood glucose (FBG) was altered and patients
with severe disease were often hyperglycemic. Data presented support
that hypoproteinaemia, hypoalbuminemia, and reduction in
high-densitylipoprotein (HDL-C) and ApoA1 were associated with disease
severity.
14.87.3 Limitations
In this study non-severe patients were divided in two groups based on
average course of the disease: mild group1 (14 days, n=28) and mild
group 2 (30 days, n=44). However mild patients with a longer disease
course did not show an intermediate phenotype (between mild patients
with shorter disease course and severe patients), hence it is unclear
whether this was a useful and how it impacted the analysis. Furthermore,
the non-exclusion of co-morbidity factors in the analysis may bias the
results (e.g. diabetic patients and glucose tests) It is not clear at
what point in time the laboratory parameters are sampled. In table 3, it
would have been interesting to explore a multivariate multiple
regression. The correlation lacks of positive control to assess the
specificity of the correlation to the disease vs. correlation in any
inflammatory case. The dynamic study assessing the predictability of the
laboratory parameter is limited to 2 patients. Hence there are several
associations with disease severity, but larger studies are necessary to
test the independent predictive value of these potential biomarkers.
14.87.4 Significance
As hospital are getting overwhelmed a set of
easily accessible laboratory indicators (such as serum total protein)
would potentially provide a triage methodology between potentially
severe cases and mild ones. This paper also opens the question regarding
metabolic deregulation and COVID-19 severity.
14.87.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.88 Viral Kinetics and Antibody Responses in Patients with COVID-19
Prospective cohort of 67 patients, clinical specimens taken and
follow-up conducted.
Viral shedding, serum IgM, IgG antibody against NP evaluated and
correlated to disease severity and clinical outcome
Viral RNA levels peaked at 1 week from febrile/cough symptom onset
in sputum, nasal swabs, and stool samples. Shedding ranged from
12-19 days (median ranges) and was longer in severe patients.
IgM and IgG titers stratified patients into three archetypes as
‘strong vs weak vs non-responders’. Strong responders (with higher
IgM/IgG titers) were significantly higher in severe patients.
14.88.3 Limitations
Specific for immune monitoring.
Not clear if stool RNA captured from live infection in
intestine/liver or from swallowed sputum. Transmission electron
microscopy (TEM) carried out on sputum samples as proof of concept,
but not stools. TEM unreasonable for actual clinical diagnosis.
Several patients had co-morbidities (such as pulmonary and liver
disease) that were not accounted for when tracking antibody
responses. Viral kinetics and IgM/IgG titers in subsets of patients
with underlying conditions/undergoing certain medication would be
informative.
14.88.4 Significance
Three archetypes of antibody response to SARS-CoV-2 with different
disease progression and kinetics is useful to stratify patients, and
for future serological tests.
Strong spike-IgG levels often correlate with lymphopenia and
CoVID-19 disease severity (2277), similar to macaque
studies in SARS (2278). It would be critical to see if anti-NP or
anti-Spike IgG antibodies for SARS-CoV-2 also elicit similar
detrimental effects before clinical use.
14.88.5 Credit
Review by Samarth Hegde as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.89 COVID-19 infection induces readily detectable morphological and inflammation-related phenotypic changes in peripheral blood monocytes, the severity of which correlate with patient outcome
This study is based on flow cytometry immunophenotyping of
PBMCs from 28 patients diagnosed positive for SARS-Cov2 (COVID19). The
authors identify a population of abnormally large (FSC-hi) monocytes,
present in COVID19 patients, but absent in PBMCs of healthy volunteers
(n=16) or patients with different infections (AIDS, malaria, TB). This
FSC-hi monocytic population contains classical, intermediate and
non-classical (monocytes (based on CD14 and CD16 expression) that
produce inflammatory cytokines (IL-6, TNF and IL-10). The authors
suggest an association of FSC-hi monocytes with poor outcome and
correlate a high percentage of FSC-low monocytes, or higher ratio of
FSC-low/hi monocytes, with faster hospital discharge.
14.89.3 Limitations
While identification of the monocytic population based
on FSC is rather robust, the characterization of these cells remains
weak. A comprehensive comparison of FSC-hi monocytes with FSC-low
monocytes from patients and healthy controls would be of high value. It
is unclear if percentages in blood are among CD45+ cells. Furthermore,
it would have been important to include absolute numbers of different
monocytic populations (in table 1 there are not enough samples and it is
unclear what the authors show).
The authors show expression of the ACE2 receptor on the surface of the
monocytes, and highlight these cells as potential targets of SARS-Cov2.
However, appropriate controls are needed. CD16 has high affinity to
rabbit IgG and it is unclear whether the authors considered unspecific
binding of rabbit anti-ACE2 to Fc receptors. Gene expression of ACE-2 on
monocytes needs to be assessed. Furthermore, it would be important to
confirm infection of monocytes by presence of viral proteins or viral
particles by microscopy.
Considering the predictive role of FSC-hi monocytes on the development
of the disease and its severity, some data expected at this level are
neither present nor addressed. Although the cohort is small, it does
include 3 ICU patients. What about their ratio of FSC-low vs FSC-hi
monocytes in comparison to other patients? Was this apparent early in
the disease course? Does this population of FSC-hi monocytes differ
between ICU patients and others in terms of frequency, phenotype or
cytokine secretion?
In general, figures need to revised to make the data clear. For example,
in Fig. 5, according to the legend it seems that patients with FSC-high
monocytes are discharged faster from the hospital. However according to
description in the text, patients were grouped in high or low levels of
FSC-low monocytes.
14.89.4 Significance
Despite the limitations of this study, the discovery of
a FSC-high monocyte population in COVID-19 patients is of great
interest. With similar implication, a the recent study by Zhou et al. (2180)
identified a connection between an inflammatory CD14+CD16+ monocyte
population and pulmonary immunopathology leading to deleterious clinical
manifestations and even acute mortality after SARS-CoV-2 infections.
Although the presence of these monocytes in the lungs has yet to be
demonstrated, such results support the importance of monocytes in the
critical inflammation observed in some COVID19 patients.
14.89.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.90 Correlation between universal BCG vaccination policy and reduced morbidity and mortality for COVID-19: an epidemiological study
The authors compared middle and high income countries that never had a
universal BCG vaccination policy (Italy, Lebanon, Nederland, Belgium)
and countries with a current policy (low income countries were excluded
from the analysis as their number of cases and deaths might be
underreported for the moment). Countries that never implement BCG
vaccination have a higher mortality rate than countries which have a BCG
vaccination policy (16.38 deaths per million people vs 0.78). Next,
the authors show that an earlier start of vaccination correlates with
a lower number of deaths per million inhabitants. They interpret this
as the vaccine protecting a larger fraction of elderly people, which are
usually more affected by COVID-19. Moreover, higher number of COVID-19
cases were presented in countries that never implemented a universal
BCG vaccination policy.
14.90.3 Limitations
While this study aims to test an intriguing hypothesis unfortunately,
the data is not sufficient at this time to accurately make any
determinations. Several caveats must be noted including: not all
countries are in the same stage of the pandemic, the number of
cases/deaths is still changing very rapidly in a lot of countries and
thus the association may only reflect exposure to the virus. This
analysis would need to be re-evaluated when all the countries are passed
the pandemic and more accurate numbers are available. Additionally, very
few middle and high-income countries ever implemented universal BCG
vaccination, which can be a source of bias (5 countries, vs 55 that have
a BCG vaccine policy). Effective screening and social isolation policies
also varied considerable across the countries tested and may reflect
another important confounder. The authors could consider analyzing the
Case Fatality Rate (CFR, % of patients with COVID-19 that die), to more
correct for exposure although testing availability will still bias this
result. Variability in mortality within countries or cities with
variable vaccination and similar exposure could also be appropriate
although confounders will still be present.
14.90.4 Significance
BCG vaccine is a live attenuated strain derived from Mycobacterium
bovis and used for a vaccine for tuberculosis (TB). This vaccine has
been proven to be efficient in preventing childhood meningitis TB, but
doesn’t prevent adult TB as efficiently. For this reason, several
countries are now only recommending this vaccine for at-risk population
only.
This study shows that there is a correlation between BCG vaccination
policy and reduced mortality for Covid-19. Indeed, BCG vaccine has
been shown to protect against several viruses and enhance innate
immunity (2281), which could explain why it could protect against SARS-CoV-2
infection, but the exact mechanism is still unknown. Moreover, the
efficiency of adult/older people vaccination and protection against
Covid-19 still needs to be assessed. Regarding this, Australian
researchers are starting a clinical trial of BCG vaccine for healthcare
workers (2282), to assess if it can protect them against Covid-19.
14.90.5 Credit
This review was undertaken as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.91 Non-neural expression of SARS-CoV-2 entry genes in the olfactory epithelium
Study analyzed bulk and scRNAseq data of olfactory cell types from
publicly-available mouse, nonhuman primate and human datasets.
show that ACE2 and TMPRSS2 (genes involved in SARS-CoV-2 entry)
are expressed in olfactory epithelial (OE) cells, basal stem cells
and respiratory epithelium (RE), but not sensory neurons.
Comparison of human RE and OE datasets (Deprez et al. 2019; Durante
et al. 2020) revealed that ACE2 and TMPRSS2 expression in OE
sustentacular cells was similar to expression in the remainder of
the non-nasal respiratory tract.
14.91.3 Limitations
Transcript data alone from healthy respiratory/olfactory cells is
not sufficient to confirm infectivity of nasal passage, or to
indicate damage to epithelia.
No mechanism defined for anosmia; it is not clear if epithelial
injury leads to reduced sensitivity or increased inflammation and
altered immune contexture drives neural/epithelial dysfunction. Will
be critical to test this in CoVID-19 patient samples or mouse
models.
14.91.4 Significance
Study provides possible rationale for anosmia observed in several
CoVID-19 patients.
Raises possibility that nasal respiratory goblet, ciliated cells,
and olfactory epithelia may serve as a viral reservoir after initial
SARS-CoV-2 infection.
Human olfactory sensory neurons express several other molecules
important to CoV (not CoV-19) entry such as FURIN, ST6GAL1,
ST3GAL4; this suggests wider mechanism of neuronal infectivity in
other coronaviruses.
14.91.5 Credit
Review by Samarth Hegde as part of a project by students, postdocs and
faculty at the Immunology Institute of the Icahn School of Medicine,
Mount Sinai.
Title:
SARS-CoV-2 proteome microarray for mapping COVID-19 antibody
interactions at amino acid resolution
This study screened the viral protein epitopes recognized by antibodies
in the serum of 10 COVID-19 patients using a new SARS-CoV-2 proteome
peptide microarray. The peptide library was constructed with 966 linear
peptides, each 15 amino acids long with a 5 amino acid overlap, based on
the protein sequences encoded by the genome of the Wuhan-Hu-1 strain.
To investigate crossreactivity between SARS-CoV-1 and SARS-CoV-2, they
tested rabbit monoclonal and polyclonal antibodies against SARS-CoV-1
nucleocapsid (N) in the microarray. Antibodies against SARS-CoV-1 N
displayed binding to the SARS-CoV-2 nucleocapsid (N) peptides.
Polyclonal antibodies showed some crossreactivity to other epitopes from
membrane (M), spike (S), ORF1ab and ORF8. This suggests that previous
exposure to SARS-CoV-1 may induced antibodies recognizing both viruses.
Screening of IgM and IgG antibodies from 10 COVID-19 patients showed
that many antibodies targeted peptides on M, N, S, Orf1ab, Orf3a, Orf7a,
and Orf8 from SARS-CoV-2, while immunodominant epitopes with antibodies
in more than 80 % COVID-19 patients were present in N, S and Orf3. It is
shown that the receptor binding domain (RBD) resides on S protein and
RBD is important for SARS-CoV-2 to enter the host cells via ACE2. Among
six epitopes on S protein, structural analysis predicted that three
epitopes were located at the surface and three epitopes were located
inside of the protein. Furthermore, some IgM antibodies from 1 patient
and IgG antibodies from 2 patients bound to the same epitope (residue
456-460, FRKSN) which resided within the RBD, and structural analysis
determined that this epitope was located in the region of the RBD loop
that engages with ACE2.
Critical analysis of the study:
In addition to the limitations mentioned in the manuscript, it would
have been informative to do the analysis over the course of the disease.
The pattern of antibody recognition, especially on S protein, and the
course of antibodies of different isotypes recognizing the same peptide
might correlate to the clinical course in these patients. It would alos
have been informative to analyze the presence of cross-reactive
antibodies from pateints previously exposed to SARS-CoV-1.
The importance and implications for the current epidemics:
This study identified linear immunodominant epitopes on SARS-CoV-2,
Wuhan-Hu-1 strain. This is a valuable information to design vaccines
that will elicit desirable immune responses.
The Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)
Directly Decimates Human Spleens and Lymph Nodes
It has been previously reported that COVID-19 patients exhibit severe
lymphocytopenia, but the mechanism through which this depletion occurs
has not been described. In order to characterize the cause and process
of lymphocyte depletion in COVID-19 patients, the authors performed
gross anatomical and in situ immune-histochemical analyses of spleens
and lymph nodes (hilar and subscapular) obtained from post-mortem
autopsies of 6 patients with confirmed positive viremia and 3 healthy
controls (deceased due to vehicle accidents).
Primary gross observations noted significant splenic and LN atrophy,
hemorrhaging, and necrosis with congestion of interstitial blood vessels
and large accumulation of mononuclear cells and massive lymphocyte
death. They found that CD68+ CD169+ cells in the spleens, hilar and
subscapular LN, and capillaries of these secondary lymphoid organs
expressed the ACE2 receptor and stain positive for the SARS-CoV-2
nucleoprotein (NP) antigen, while CD3+ T cells and B220+ B cells
lacked both the ACE2 receptor and SARS-CoV-2 NP antigen. ACE2+ NP+
CD169+ macrophages were positioned in the splenic marginal zone (MZ)
and in the marginal sinuses of LN, which suggests that these macrophages
were positioned to encounter invading pathogens first and may contribute
to virus dissemination.
Since SARS-CoV-2 does not directly infect lymphocytes, the authors
hypothesized that the NP+ CD169+ macrophages are responsible for
persistent activation of lymphocytes via Fas::FasL interactions that
would mediate activation-induced cell death (AICD). Indeed, the
expression of Fas was significantly higher in virus-infected tissue than
that of healthy controls, and TUNEL staining showed significant
lymphocytic apoptosis. Since pro-inflammatory cytokines like IL-6 and
TNF-α can also engage cellular apoptosis and necrosis, the authors
interrogated the cytokine expression of the secondary lymphoid organs
from COVID-19 patients; IL-6, not TNF-α, was elevated in virus-infected
splenic and lymph node tissues, compared to those of healthy controls,
and immunofluorescent staining showed that IL-6 is primarily produced by
the infected macrophages. In vitro infection of THP1 cells with
SARS-CoV-2 spike protein resulted in selectively increased Il6
expression, as opposed to Il1b and Tnfa transcription. Collectively,
the authors concluded that a combination of Fas up-regulation and IL-6
production by NP+ CD169+ macrophages induce AICD in lymphocytes in
secondary lymphoid organs, resulting in lymphocytopenia.
In summary, this study reports that CD169+ macrophages in the splenic
MZ, subscapular LN, and the lining capillaries of the secondary lymphoid
tissues express ACE2 and are susceptible to SARS-CoV-2 infection. The
findings point to the potential role of these macrophages in viral
dissemination, immunopathology of these secondary lymphoid organs,
hyperinflammation and lymphopenia.
Limitations
Technical
A notable technical limitation is the small number of samples (n=6);
moreover, the analysis of these samples using multiplexed
immunohistochemistry and immunofluorescence do not necessarily provide
the depth of unbiased interrogation needed to better identify the cell
types involved.
Biological
The available literature and ongoing unpublished studies, including
single-cell experiments of spleen and LN from organ donors, do not
indicate that ACE2 is expressed by macrophages; however, it remains
possible that ACE2 expression may be triggered by type I IFN in COVID-19
patients. Importantly, the SARS-CoV-2 NP staining of the macrophages
does not necessarily reflect direct infection of these macrophages;
instead, positive staining only indicates that these macrophages carry
SARS-CoV-2 NP as antigen cargo, which may have been phagocytosed. Direct
viral culture of macrophages isolated from the secondary lymphoid organs
with SARS-CoV-2 is required to confirm the potential for direct
infection of macrophages by SARS-CoV-2. Additionally, it is important to
note that the low to negligible viremia reported in COVID-19 patients
to-date does not favor a dissemination route via the blood, as suggested
by this study, which would be necessary to explain the presence of
virally infected cells in the spleen.
Relevance
Excess inflammation in response to SARS-CoV-2 infection is characterized
by cytokine storm in many COVID-19 patients. The contribution of this
pathology to the overall fatality rate due to COVID-19, not even
necessarily directly due to SARS-CoV-2 infection, is significant. A
better understanding of the full effect and source of some of these
major cytokines, like IL-6, as well as the deficient immune responses,
like lymphocytopenia, is urgently needed. In this study, the authors
report severe tissue damage in spleens and lymph nodes of COVID-19
patients and identify the role that CD169+ macrophages may play in the
hyperinflammation and lymphocytopenia that are both characteristic of
the disease. It may, therefore, be important to note the effects that
IL-6 inhibitors like Tocilizumab and Sarilumab may specifically have on
splenic and LN function. It is important to note that similar
observations of severe splenic and LN necrosis and inflammation in
patients infected with SARS-CoV-1 further support the potential
importance and relevance of this study.
14.92 Cigarette smoke triggers the expansion of a subpopulation of respiratory epithelial cells that express the SARS-CoV-2 receptor ACE2
Study uses scRNAseq, bulk seq data and air-liquid interface culture
experiments to show that cigarette smoke causes a dose-dependent
upregulation of ACE2 in mouse and human lungs (transplantation,
tumor resection, or IPF datasets).
ACE2 was not up-regulated in patients with asthma or
lung-sarcoidosis or in mouse models of cystic fibrosis or carcinogen
exposure.
Cathepsin B (alternate protease involved in viral entry) is
increased in smoke-exposed mouse or human lungs.
Smoke triggers a protective expansion of mucus-secreting MUC5AC+
goblet and SCGB1A1+ club cells; ACE2 presence in these cells is
increased upon smoke exposure.
14.92.3 Limitations:
Long-term smokers usually have several co-morbidities including
immune dysfunction, which can affect interpretation of CoV-2
susceptibility in these datasets. Ideally, analyses can control for
major co-morbidities across smokers and non-smokers (immune
suppression, cardiovascular disease and atherosclerosis).
Hyperplasia of ACE2+ goblet cells upon smoking needs to be separated
from ACE2 upregulation in existing goblet cells.
ACE2 expression increase alone does not confirm increased viral
entry into goblet cells; future studies with air-liquid interface
cultures testing CoV-2 infectivity in ex vivo epithelial cells
from human epithelial lines, ex vivo samples or hACE2 mice will be
very informative.
14.92.4 Significance
This study may partially explain why smokers are more likely to
develop severe SARS-CoV-2 infections. Also, the reversibility of
ACE2 expression upon smoking cessation suggests that quitting
smoking could lessen CoV-2 susceptibility.
Absence of ACE2 upregulation in other lung inflammation pathologies
implies CoV-2 susceptibility might be smoking-specific, and not
fibrosis-specific.
Another preprint showed ACE2 expression increases in lung of
patients with CoV-2 co-morbidities such as hypertension (2257); these studies
collectively paint a better picture of CoV-2 susceptibility before
actual experiments can be carried out.
14.92.5 Credit
Review by Samarth Hegde as part of a project by students, postdocs and faculty at the Immunology Institute of the Icahn School of Medicine, Mount Sinai.
14.93 The comparative superiority of IgM-IgG antibody test to real-time reverse transcriptase PCR detection for SARS-CoV-2 infection diagnosis
The study compares IgM and IgG antibody testing to RT-PCR detection of
SARS-CoV-2 infection. 133 patients diagnosed with SARS-CoV-2 in Renmin
Hospital (Wuhan University, China) were analyzed. The positive ratio was
78.95% (105/133) in IgM antibody test (SARS-CoV-2 antibody detection kit
from YHLO Biotech) and 68.42% (91/133) in RT-PCR (SARS-CoV-2 ORF1ab/N
qPCR detection kit). There were no differences in the sensitivity of
SARS-CO-V2 diagnosis in patients grouped according to disease severity.
For example, IgG responses were detected in 93.18% of moderate cases,
100% of severe cases and 97.3% of critical cases. In sum, positive
ratios were higher in antibody testing compared to RT-PCR detection,
demonstrating a higher detection sensitivity of IgM-IgG testing for
patients hospitalized with COVID-19 symptoms.
14.93.3 Limitations
This analysis only included one-time point of 133 hospitalized patients,
and the time from symptom onset was not described. There was no
discussion about specificity of the tests and no healthy controls were
included. It would be important to perform similar studies with more
patients, including younger age groups and patients with mild symptoms
as well as asymptomatic individuals. It is critical to determine how
early after infection/symptom onset antibodies can be detected and the
duration of this immune response.
14.93.4 Significance
The IgM-IgG combined testing is important to improve clinical
sensitivity and diagnose COVID-19 patients. The combined antibody test
shows higher sensitivity than individual IgM and IgG tests or nucleic
acid-based methods, at least in patients hospitalized with symptoms.
14.93.5 Credit
Review by Erica Dalla as part of a project by students, postdocs and
faculty at the Immunology Institute of the Icahn School of Medicine,
Mount Sinai.
Title: Lectin-like Intestinal Defensin Inhibits 2019-nCoV Spike
binding to ACE2
Human ACE2 was previously identified as the host receptor for
SARS-CoV-2. Despite ACE2 being expressed in both lung alveolar
epithelial cells and small intestine enterocytes, respiratory problems
are the most common symptom after viral infection while intestinal
symptoms are much less frequent. Thus, the authors here investigate the
biology behind the observed protection of the intestinal epithelium from
SARS-CoV-2. Human defensin 5 (HD5), produced by Paneth cells in the
small intestine, was shown to interact with human ACE2, with a binding
affinity of 39.3 nM by biolayer interferometry (BLI). A blocking
experiment using different doses of HD5 coating ACE2 showed that HD5
lowered viral spike protein S1 binding to ACE2. Further, a molecular
dynamic simulation demonstrated a strong intermolecular interaction
between HD5 and the ACE2 ligand binding domain. To test HD5 inhibitory
effect on S1 binding to ACE2, human intestinal epithelium Caco-2 cells
were preincubated with HD5. Preincubation strongly reduced adherence of
S1 to surface of cells. HD5 was effective at a concentration as low as
10 µg/mL, comparable to the concentration found in the intestinal fluid.
Limitations:
The study focuses exclusively on intestinal cells. However, HD5 could
have been tested to block ACE2-S1 binding in human lung epithelial cells
as a potential treatment strategy. It would be useful to know whether
HD5 could also prevent viral entry in lung cells.
Relevance:
This work provides the first understanding of the different efficiency
of viral entry and infection among ACE2-expressing cells and tissues.
Specifically, the authors show that human defensin 5 produced in the
small intestine is able to block binding between S1 and ACE2 necessary
for viral entry into cells. The study provides a plausible explanation
on why few patients show intestinal symptoms and suggests that patients
with intestinal disease that decrease defensins’ production may be more
susceptible to SARS-CoV-2. It also indicates that HD5 could be used as a
molecule to be exogenously administered to patients to prevent viral
infection in lung epithelial cells.
Title:
Susceptibility of ferrets, cats, dogs and different domestic animals
to SARS-coronavirus-2
This study evaluated the susceptibility of different model laboratory
animals (ferrets), as well as companion (cats and dogs), and domestic
animals (pigs, chickens and ducks) to SARS-CoV-2. They tested infection
with two SARS-CoV2 isolates, one from an environmental sample collected
in the Huanan Seafood Market in Wuhan (F13-E) and the other from a human
patient in Wuhan (CTan-H).
Ferrets were inoculated with either of the two viruses by intranasal
route with 105 pfu, and the viral replication was evaluated. Two
ferrets from each group were euthanized on day 4 post infection (p.i.).
AT day 4 p.i., viral RNA and infectious viruses were detected only in
upper respiratory tract (nasal turbinate, upper palate, tonisls, but not
in the trachea, lungs or other tissues. Viral RNA and virus titer in the
remaining ferrets were monitored in nasal washes and rectal swabs on
days 2, 4, 6, 8 and 10 p.i. Viral RNA and infectious viruses were
detected in nasal washes until day 8 p.i. One ferret in each group
developed fever and loss of appetite on days 10 and 12 p.i., however,
viral RNA was practically undetactable. These two ferrets showed severe
lymphoplasmacytic perivasculitis and vasculitis in the lungs and lower
antibody titers compare to other 4 ferrets.
Cats. Five subadult 8-month-old domestic cats were inoculated with
CTan-h virus and three uninfected cats were placed in a cage adjacent to
each of the infected cats to monitor respiratory droplet transmission.
Viral RNA was detected in the upper respiratory organs from all infected
cats and in one out of three exposed cats. All infected (inoculated and
exposed) cats developed elevated antibodies against SARS-CoV2. Viral
replication studies with juvenile cats (70-100 days) revealed massive
lesions in the nasal and tracheal mucosa epithelium and lungs of two
inoculated cats which died or were euthanized on day 3 p.i., and
infection in one out of three exposed cats. These results indicated
SARS-CoV2 could replicate in cats, that juvenile cats were more
susceptible that adults, and theat SARS-CoV2 could be transmit via
respiratory droplets between cats.
Dogs and others. Five 3-month-old beagle dogs were inoculated and
housed with two uninoculated beagles in a room. Two virus inoculated
dogs seroconverted, but others including two contact dogs were all
seronegative for SARS-CoV2 and infectious virus was not detected in any
swabs collected. Viral RNA was not detected in swabs from pigs,
chickens, and ducks inoculated or contacted. These results indicated
that dogs, pigs, chickens, and ducks might have low or no susceptibility
to SARS-CoV2.
Critical analysis of the study:
This manuscript describes the viral replication and clinical symptoms of
SARS-CoV2 infection in ferrets, and the SARS-CoV2 infection and
transmission in cats. Clinical and pathological analysis was not
performed in cats, therefore the correlation of virus titer with
symptoms severity in the adult and juvenile cats could not be
determined.
The importance and implications for the current epidemics:
SARS-CoV-2 transmission to tigers, cats and dogs has been previously
reported. It should be noted that this manuscript did not evaluate the
transmission from cats to human. Nevertheless, it clearly showed higher
susceptibility of ferrets and domestic cats to SARS-CoV-2. This data
strongly indicates the need for surveillance of possible infection and
transmission of SARS-CoV-2 by domestic cats.
14.94 Virus-host interactome and proteomic survey of PMBCs from COVID-19 patients reveal potential virulence factors influencing SARS-CoV-2 pathogenesis
The authors identified intra-viral protein-protein interactions
(PPI) with two different approaches: genome wide yeast-two hybrid (Y2H)
and co-immunoprecipitation (co-IP). A total of 58 distinct PPI were
characterized. A screen of viral-host PPI was also established by
overexpressing all the SARS-CoV-2 genes with a Flag epitope into HEK293
cells and purifying each protein complex. Interacting host proteins were
then identified by liquid chromatography and tandem mass spectrometry.
251 cellular proteins were identified, such as subunits of ATPase, 40S
ribosomal proteins, T complex proteins and proteasome related proteins,
for a total of 631 viral-host PPI. Several interactions suggesting
protein-mediated modulation of the immune response were identified,
highlighting the multiple ways SARS-CoV-2 might reprogram infected
cells.
Subsequently, the authors compared global proteome profiles of PBMCs
from healthy donors (n=6) with PBMC from COVID-19 patients with mild
(n=22) or severe (n=13) symptoms. 220 proteins were found to be
differentially expressed between healthy donors and mild COVID-19
patients, and a pathway analysis showed a general activation of the
innate immune response. 553 proteins were differentially expressed
between the PBMC of mild and severe COVID-19 patients, most of them
(95%) being downregulated in severe patients. Functional pathway
analysis indicated a defect of T cell activation and function in severe
COVID-19. There was also evidence suggesting reduced antibody secretion
by B cells. Together, these results suggest a functional decline of
adaptive immunity. A FACS analysis of PBMC from severe patients
indicated higher levels of IL6 and IL8 but not IL17 compared to mild
patients.
Finally, the authors focused on NKRF, an endogenous repressor of IL8/IL6
synthesis that was previously identified as interacting with SARS-Cov-2
nsp9,10,12,13 and 15. Individually expressed nsp9 and nsp10 (but not
nsp12, nsp13, nsp15) induced both IL6 and IL8 in lung epithelial A459
cells, indicating that nsp9 and nsp10 may be directly involved in the
induction of these pro-inflammatory cytokines. The authors finally argue
that nsp9 and nsp10 represent potential drug targets to prevent
over-production of IL6 and IL8 in infected cells, and reducing the
over-activation of neutrophils.
14.94.3 Limitations
First, the authors seem to have forgotten to include the extended data
in the manuscript, and their proteomic data does not seem to be publicly
available for the moment, which limits greatly our analysis of their
results.
While this work provides important data on host and viral PPI, only 19
interactions were identified by Y2H system but 52 with co-IP. The
authors do not comment about what could lead to such differences between
the two techniques and they don’t specify whether they detected the same
interactions using the two techniques.
Moreover, the PBMC protein quantification was performed comparing bulk
PBMC. Consequently, protein differences likely reflect differences in
cell populations rather than cell-intrinsic differences in protein
expression. While this analysis is still interesting, a similar
experiment performed on pre-sorted specific cell populations would allow
measuring proteome dynamics at a higher resolution.
Finally, the authors did not discussed their results in regards to
another SARS-CoV-2 interactome of host-viral PPI that had been published
previously1. This study reported 332 host-virus PPI, but no
interaction of viral proteins with NKRF was found. Some interactions
were found in both studies (eg. N and G3BP1, Orf6 and RAE1). However,
the time point used to lyse the cells were different (40h previously vs
72h here), which could explain some of the differences.
14.94.4 Relevance
The identification of many interactions between intra-viral and
host-virus PPI provides an overview of host protein and pathways that
are modulated by SARS-CoV-2, which can lead to the identification of
potential targets for drug development.
In the model proposed by the authors, nsp9 and nsp10 from SARS-Cov-2
induce an over-expression of IL6 and IL8 by lung epithelial cells, which
recruits neutrophils and could lead to an excess in lung infiltration.
Nsp9 has been shown to be essential for viral replication for
SARS-Cov-12, and shares a 97% homology with nsp9 from SARS-Cov-23.
Further, nsp9 crystal structure was recently solved3, which can help
to develop drug inhibitors if this protein is further confirmed as being
important for the virulence of SARS-Cov-2.
1. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS-CoV-2-Human
Protein-Protein Interaction Map Reveals Drug Targets and Potential
Drug-Repurposing. bioRxiv. March 2020:2020.03.22.002386.
doi:10.1101/2020.03.22.002386
2. Miknis ZJ, Donaldson EF, Umland TC, Rimmer RA, Baric RS, Schultz LW.
Severe acute respiratory syndrome coronavirus nsp9 dimerization is
essential for efficient viral growth. J Virol. 2009;83(7):3007-3018.
doi:10.1128/JVI.01505-08
3. Littler DR, Gully BS, Colson RN, Rossjohn J. Crystal Structure of
the SARS-CoV-2 Non-Structural Protein 9, Nsp9. Molecular Biology; 2020.
doi:10.1101/2020.03.28.013920
Title: Prediction and Evolution of B Cell Epitopes of Surface
Protein in SARS-CoV-2
Lon et al. used a bioinformatic analysis of the published SARS-CoV-2
genomes in order to identify conserved linear and conformational B cell
epitopes found on the spike (S), envelope (E), and membrane (M)
proteins. The characterization of the surface proteins in this study
began with an assessment of the peptide sequences in order to identify
hydrophilicity indices and protein instability indices using the
Port-Param tool in ExPASy. All three surface proteins were calculated to
have an instability score under 40 indicating that they were stable.
Linear epitopes were identified on the basis of surface probability and
antigenicity, excluding regions of glycosylation. Using BepiPred 2.0
(with a cutoff value of 0.35) and ABCpred (with a cutoff value of 0.51),
4 linear B cell epitopes were predicted for the S protein, 1 epitope for
the E protein, and 1 epitope for the M protein. For structural analysis,
SARS-CoV assemblies published in the Protein Data Bank (PDB) acting as
scaffolds for the SARS-CoV-2 S and E amino acid sequences were used for
input into the SWISS-MODEL server in order to generate three-dimensional
structural models for the assessment of conformational epitopes. Using
Ellipro (cutoff value of 0.063) and SEPPA (cutoff value of 0.5), 1
conformational epitope was identified for the S protein and 1 epitope
was identified for the E protein, both of which are accessible on the
surface of the virus. Finally, the Consurf Server was used to assess the
conservation of these epitopes. All epitopes were conserved across the
published SARS-CoV-2 genomes and one epitope of the spike protein was
predicted to be the most stable across coronavirus phylogeny.
Critical Analysis/Limitations:
While this study provides a preliminary identification of potential
linear and conformational B cell epitopes, the translational value of
the epitopes described still needs extensive experimental validation to
ascertain whether these elicit a humoral immune response. The
conformational epitope analyses are also limited by the fact that they
are based off of predicted 3D structure from homology comparisons and
not direct crystal structures of the proteins themselves. Additionally,
since there was not a published M protein with a high homology to
SARS-CoV-2, no conformational epitopes were assessed for this protein.
Finally, while evolutionary conservation is an important consideration
in understanding the biology of the virus, conservation does not
necessarily imply that these sites neutralize the virus or aid in
non-neutralizing in vivo protection.
Relevance/Implications:
With further experimental validation that confirms that these epitopes
induce effective antibody responses to the virus, the epitopes described
can be used for the development of treatments and vaccines as well as
better characterize the viral structure to more deeply understand
pathogenesis.
15 Appendix B
Contributors were asked to complete this template to summarize and evaluate new papers related to diagnostics.
Title: Please edit the title to add the name of the paper after the colon
Please paste a link to the paper or a citation here:
Please list some keywords (3-10) that help identify the relevance of this paper to COVID-19
keyword 1 (replace me, copy and paste more than three if needed)
keyword 2 (replace me, copy and paste more than three if needed)
keyword 3 (replace me, copy and paste more than three if needed)
Please note the publication / review status
Pre-print
New Peer-Reviewed Paper
Peer-Reviewed Paper Pre-2020
Which areas of expertise are particularly relevant to the paper?
virology
epidemiology
biostatistics
immunology
pharmacology
Questions to answer about each paper:
Please provide 1-2 sentences introducing the study and its main findings
Study question(s) being investigated:
What type of testing scenario is being considered?
Is it a screening test (used for individuals with no symptoms), diagnostic test (used for individuals with symptoms), or definitive test (used for individuals who have had previous positive test results on diagnostic or screening tests)?
Study population:
What is the model system (e.g., human study, animal model, cell line study)?
What is the sample size?
What is the “pre-test” probability of disease in the study population (i.e., what is the anticipated prevalence of the disease?)
For human studies, the following are related to the pre-test probability:
What countries/regions are considered?
What is the age range, gender, other relevant characteristics?
What is the setting of the study (e.g., random sample of school children, retirement communities, etc.)?
What other specific inclusion-exclusion criteria are considered?
Reference test:
What reference test is considered as a “gold standard” comparator for the test under investigation?
Test assignment:
How are the new and reference tests assigned?
Examples of assignment could include: Recruited individuals have initially undergone neither the new nor the reference test; individuals tested as positive or negative by the reference test undergo the new test; individuals who have undertaken the new test are assessed by the standard test.
Are there any other relevant details about the study design?
Depending on how individuals are chosen, the test may be biasing towards more sick or less sick individuals or very clear-cut positive/negative cases.
Any factors that would influence this bias should be included here.
Test conduct:
How were tests performed?
Describe technical details of assays used, when measurements were taken and by whom, etc. for both the new and standard tests.
Test Assessment
Describe how individuals are classified as positive or negative, e.g. if a threshold is used.
Is there evidence that the test is precise/reproducible when repeated more than once?
Are measurements complete?
For example: Do some participants undergo just one test (the new or the reference test)?
Are there individuals with inconclusive results?
Results summary:
What are the estimated sensitivity, specificity, positive predictive value (PPV), and negative predicted value (NPV)?
Note that the PPV and NPV represent “post-test” probabilities of disease and are generally more meaningful than sensitivity and specificity.
Sometimes the post-test odds will be given instead.
What are the confidence bounds around these intervals?
Interpretation of results for study population:
How good is the test at ruling in or ruling out a disease based on the post-test probabilities?
Are there identified side affects of the test?
Is patient adherence to the test likely to be an issue?
Extrapolation of conclusions to other groups of individuals
How well is the test likely to work in populations with different pretest odds?
For example, if the prevalence is lower, then the PPV will also be lower, but the NPV will be higher.
How costly is the test?
How difficult is it to perform the test in different settings?
Could the test be combined with other existing tests?
Summary of reliability
1-2 sentences on concluding remarks, including summary of strengths, weaknesses, limitations.
Progress
Check off the components as they are completed. If the component is not applicable, check the box as well.
1-2 sentences introducing the study and its main findings
Describe testing scenario
Describe model system
Sample size
Describe prevalnce of disease
Describe countries/regions are considered
Describe age range, gender, other relevant characteristics
Describe setting of the study
Describe other specific inclusion-exclusion criteria
Describe “gold standard”
Describe how the new and reference tests assigned
Describe other relevant details about the study design
Describe how the tests were performed
Describe how individuals are classified as positive or negative
Describe if test is precise/reproducible
Describe whether measurements are complete
What are the estimated sensitivity, specificity, positive predictive value (PPV), and negative predicted value (NPV)?
What are the confidence bounds around these intervals?
Describe post-test probabilities
Describe side affects of the test
Describe patient adherence
Describe how it will extrapolate
How costly is the test?
How difficult is it to perform the test in different settings?
Could the test be combined with other existing tests?
Summary of reliability
16 Appendix C
Contributors were asked to complete this template to summarize and evaluate new papers related to therapeutics.
Title: Please edit the title to add the name of the paper after the colon
Please paste a link to the paper or a citation here:
Please list some keywords (3-10) that help identify the relevance of this paper to COVID-19
keyword 1 (replace me, copy and paste more than three if needed)
keyword 2 (replace me, copy and paste more than three if needed)
keyword 3 (replace me, copy and paste more than three if needed)
Please note the publication / review status
Pre-print
New Peer-Reviewed Paper
Peer-Reviewed Paper Pre-2020
Which areas of expertise are particularly relevant to the paper?
virology
epidemiology
biostatistics
immunology
pharmacology
Questions to answer about each paper:
Please provide 1-2 sentences introducing the study and its main findings
Study question(s) being investigated:
How many/what drugs/combinations are being considered?
What are the main hypotheses being tested?
Study population:
What is the model system (e.g., human study, animal model, cell line study)?
What is the sample size? If multiple groups are considered, give sample size for each group (including controls).
number treated with treatment A
number treated with treatment B
For human studies:
What countries/regions are considered?
What is the age range, gender, other relevant characteristics?
What is the setting of the study (random sample of school children, inpatient, outpatient, etc)?
What other specific inclusion-exclusion criteria are considered?
For example, do the investigators exclude patients with diagnosed neoplasms or patients over/under a certain age?
Treatment assignment:
How are treatments assigned?
For example, is it an interventional or an observational study?
Is the study randomized?
A study can be interventional but not randomized (e.g., a phase I or II clinical trial is interventional but often not randomized).
Provide other relevant details about the design.
This includes possible treatment stratification (e.g., within litters for animal studies, within hospitals for human studies), possible confounding variables (e.g., having a large age range of individuals), possible risks of bias and how they are addressed (e.g., is there masking in a clinical trial? how are individuals chosen in an observational study?).
Outcome Assessment:
Describe the outcome that is assessed and whether it is appropriate.
For example: Is the outcome assessed by a clinician or is it self-reported?
Is the outcome based on viral load or a functional measurement (e.g., respiratory function, discharge from hospital)?
What method is used to measure the outcome?
How long after a treatment is the outcome measured?
Are outcome measurements complete?
For example, are there individuals lost to follow up?
Are outcome measurements subject to various kinds of bias?
For example, a lack of masking in randomized clinical trials.
Statistical Methods Assessment:
What methods are used for inference?
For example, logistic regression, nonparametric methods.
Are the methods appropriate for the study?
For example, are clustered data treated independently or are clusters adjusted for, such as different hospitals or litters?
Are adjustments made for possible confounders?
For example, adjustment for age, sex, or comorbidities.
Results Summary:
What is the estimated association?
For example, is it an estimated odds ratio, a median difference in detected cases, etc?
What measures of confidence or statistical significance are provided?
For example, confidence intervals, p-values, and/or Bayes factors.
Interpretation of results for study population:
Can we make a causal interpretation for the individuals in the study of drug -> outcome, such as “taking drug A improves likelihood of survival twofold over taking drug B.”
For example, with a well-performed animal study or randomized trial it is often possible to infer causality.
If is an observational study, does it match up with some of the Bradford Hill criteria? https://www.edwardtufte.com/tufte/hill https://en.wikipedia.org/wiki/Bradford_Hill_criteria
Are there identified side effects or interactions with other drugs?
For example, is the treatment known to cause liver damage or to not be prescribed for individuals with certain comorbities?
Are there specific subgroups with different findings?
For example, do individuals with a specific baseline seem to do particularly well? Be particularly cautious with respect to multiple testing here.
Extrapolation of conclusions to other groups of individuals not specifically included in the study:
If the study is an animal study, which animal and how relevant is that model?
Is the model system appropriate? Is there evidence from past use that it’s highly-relevant to therapeutic design in this context?
If it is a human study, what characteristics of the study population may support/limit extrapolation?
Can results extrapolate easily to other similar groups? (e.g., same country, similar age groups)
What would happen if conditions are extended in terms of dose or duration?
Can results be extrapolated to other populations or in very different settings? (e.g., different age group, primary care setting vs emergency department etc)
Summary of reliability
1-2 sentences on concluding remarks, including summary of strengths, weaknesses, limitations.
Progress
Check off the components as they are completed. If the component is not applicable, check the box as well.
1-2 sentences introducing the study and its main findings
Describe How many/what drugs/combinations are being considered
Describe the model system
What is the sample size?
What countries/regions are considered
What is the age range, gender, other relevant characteristics
Describe study setting
Describe other specific inclusion-exclusion criteria
Describe how treatments are assigned
Describe randomization (or not) and other relavent details about the design
Describe the outcome that is assessed and whether it is appropriate.
Describe whether the outcome measurements are complete
Are outcome measurements subject to various kinds of bias?
Describe methods used for inference
Describe whether the methods are appropriate for the study
Are adjustments made for possible confounders?
Describe the estimated association
What measures of confidence or statistical significance are provided?
Describe whether a causal interpretation can be made
Are there identified side effects or interactions with other drugs?
Are there specific subgroups with different findings?
If the study is an animal study, which animal and how relevant is that model?
If it is a human study, what characteristics of the study population may support/limit extrapolation?
Summary of reliability
17 Appendix D
Contributors were asked to complete this template to summarize and evaluate new papers related to topics besides therapeutics and diagnostics.
Title: Please edit the title to add the name of the paper after the colon.
General Information
Please paste a link to the paper or a citation here:
Is this paper primarily relevant to Background or Pathogenesis?
Background
Pathogenesis
Methods
Please list some keywords (3-10) that help identify the relevance of this paper to COVID-19
keyword 1 (replace me, copy and paste more than three if needed)
keyword 2 (replace me, copy and paste more than three if needed)
keyword 3 (replace me, copy and paste more than three if needed)
Please note the publication / review status
Pre-print
New Peer-Reviewed Paper
Peer-Reviewed Paper Pre-2020
Which areas of expertise are particularly relevant to the paper?
virology
epidemiology
biostatistics
immunology
pharmacology
other:
Summary
Suggested questions to answer about each paper:
- What did they analyze?
- What methods did they use?
- Does this paper study COVID-19, SARS-CoV-2, or a related disease and/or virus?
- What is the main finding (or a few main takeaways)?
- What does this paper tell us about the background and/or diagnostics/therapeutics for COVID-19 / SARS-CoV-2?
- Do you have any concerns about methodology or the interpretation of these results beyond this analysis?