This manuscript (permalink) was automatically generated from greenelab/deep-review@44ff95a on December 28, 2021.
![]() Brock C. Christensen2.1⚄,
Brock C. Christensen2.1⚄,
![]() Anthony Gitter2.2,2.3⚄,†,
Anthony Gitter2.2,2.3⚄,†,
![]() Daniel S. Himmelstein2.4⚄,
Daniel S. Himmelstein2.4⚄,
![]() Alexander J. Titus2.1⚄,
Alexander J. Titus2.1⚄,
![]() Joshua J. Levy2.5⚄,
Joshua J. Levy2.5⚄,
![]() Casey S. Greene2.6,2.7⚄,†,
Casey S. Greene2.6,2.7⚄,†,
![]() Daniel C. Elton2.8⚄,
The Version 1.0 Deep Review Authors
Daniel C. Elton2.8⚄,
The Version 1.0 Deep Review Authors
⚄ — Author order for version 2.0 is currently randomized with each new build.
† — To whom correspondence should be addressed: gitter@biostat.wisc.edu (A.G.) and greenescientist@gmail.com (C.S.G.)
2.1. Department of Epidemiology, Geisel School of Medicine, Dartmouth College, Lebanon, NH
2.2. Department of Biostatistics and Medical Informatics, University of Wisconsin-Madison, Madison, WI
2.3. Morgridge Institute for Research, Madison, WI
2.4. Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America
2.5. Program in Quantitative Biomedical Sciences, Geisel School of Medicine at Dartmouth, Lebanon, NH
2.6. Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA
2.7. Childhood Cancer Data Lab, Alex’s Lemonade Stand Foundation, Philadelphia, PA
2.8. Radiology and Imaging Sciences, National Institutes of Health Clinical Center, Bethesda, MD
![]() Travers Ching1.1,☯,
Travers Ching1.1,☯,
![]() Daniel S. Himmelstein1.2,
Daniel S. Himmelstein1.2,
![]() Brett K. Beaulieu-Jones1.3,
Brett K. Beaulieu-Jones1.3,
![]() Alexandr A. Kalinin1.4,
Alexandr A. Kalinin1.4,
![]() Brian T. Do1.5,
Brian T. Do1.5,
![]() Gregory P. Way1.2,
Gregory P. Way1.2,
![]() Enrico Ferrero1.8,
Enrico Ferrero1.8,
![]() Paul-Michael Agapow1.9,
Paul-Michael Agapow1.9,
![]() Michael Zietz1.2,
Michael Zietz1.2,
![]() Michael M. Hoffman1.10,1.11,1.12,
Michael M. Hoffman1.10,1.11,1.12,
![]() Wei Xie1.13,
Wei Xie1.13,
![]() Gail L. Rosen1.14,
Gail L. Rosen1.14,
![]() Benjamin J. Lengerich1.15,
Benjamin J. Lengerich1.15,
![]() Johnny Israeli1.16,
Johnny Israeli1.16,
![]() Jack Lanchantin1.17,
Jack Lanchantin1.17,
![]() Stephen Woloszynek1.14,
Stephen Woloszynek1.14,
![]() Anne E. Carpenter1.18,
Anne E. Carpenter1.18,
![]() Avanti Shrikumar1.19,
Avanti Shrikumar1.19,
![]() Jinbo Xu1.20,
Jinbo Xu1.20,
![]() Evan M. Cofer1.21,1.22,
Evan M. Cofer1.21,1.22,
![]() Christopher A. Lavender1.23,
Christopher A. Lavender1.23,
![]() Srinivas C. Turaga1.24,
Srinivas C. Turaga1.24,
![]() Amr M. Alexandari1.19,
Amr M. Alexandari1.19,
![]() Zhiyong Lu1.25,
Zhiyong Lu1.25,
![]() David J. Harris1.26,
David J. Harris1.26,
![]() Dave DeCaprio1.27,
Dave DeCaprio1.27,
![]() Yanjun Qi1.17,
Yanjun Qi1.17,
![]() Anshul Kundaje1.19,1.28,
Anshul Kundaje1.19,1.28,
![]() Yifan Peng1.25,
Yifan Peng1.25,
![]() Laura K. Wiley1.29,
Laura K. Wiley1.29,
![]() Marwin H.S. Segler1.30,
Marwin H.S. Segler1.30,
![]() Simina M. Boca1.31,
Simina M. Boca1.31,
![]() S. Joshua Swamidass1.32,
S. Joshua Swamidass1.32,
![]() Austin Huang1.33,
Austin Huang1.33,
![]() Anthony Gitter1.34,1.35,†,
Anthony Gitter1.34,1.35,†,
![]() Casey S. Greene1.2,†
Casey S. Greene1.2,†
☯ — Author order was determined with a randomized algorithm
† — To whom correspondence should be addressed: gitter@biostat.wisc.edu (A.G.) and greenescientist@gmail.com (C.S.G.)
1.1. Molecular Biosciences and Bioengineering Graduate Program, University of Hawaii at Manoa, Honolulu, HI
1.2. Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA
1.3. Genomics and Computational Biology Graduate Group, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA
1.4. Department of Computational Medicine and Bioinformatics, University of Michigan Medical School, Ann Arbor, MI
1.5. Harvard Medical School, Boston, MA
1.6. Program in Quantitative Biomedical Sciences, Geisel School of Medicine at Dartmouth, Lebanon, NH
1.7. Department of Epidemiology, Geisel School of Medicine, Dartmouth College, Lebanon, NH
1.8. Computational Biology and Stats, Target Sciences, GlaxoSmithKline, Stevenage, United Kingdom
1.9. Data Science Institute, Imperial College London, London, United Kingdom
1.10. Princess Margaret Cancer Centre, Toronto, ON, Canada
1.11. Department of Medical Biophysics, University of Toronto, Toronto, ON, Canada
1.12. Department of Computer Science, University of Toronto, Toronto, ON, Canada
1.13. Electrical Engineering and Computer Science, Vanderbilt University, Nashville, TN
1.14. Ecological and Evolutionary Signal-processing and Informatics Laboratory, Department of Electrical and Computer Engineering, Drexel University, Philadelphia, PA
1.15. Computational Biology Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA
1.16. Biophysics Program, Stanford University, Stanford, CA
1.17. Department of Computer Science, University of Virginia, Charlottesville, VA
1.18. Imaging Platform, Broad Institute of Harvard and MIT, Cambridge, MA
1.19. Department of Computer Science, Stanford University, Stanford, CA
1.20. Toyota Technological Institute at Chicago, Chicago, IL
1.21. Department of Computer Science, Trinity University, San Antonio, TX
1.22. Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, NJ
1.23. Integrative Bioinformatics, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC
1.24. Howard Hughes Medical Institute, Janelia Research Campus, Ashburn, VA
1.25. National Center for Biotechnology Information and National Library of Medicine, National Institutes of Health, Bethesda, MD
1.26. Department of Wildlife Ecology and Conservation, University of Florida, Gainesville, FL
1.27. ClosedLoop.ai, Austin, TX
1.28. Department of Genetics, Stanford University, Stanford, CA
1.29. Division of Biomedical Informatics and Personalized Medicine, University of Colorado School of Medicine, Aurora, CO
1.30. Institute of Organic Chemistry, Westfälische Wilhelms-Universität Münster, Münster, Germany
1.31. Innovation Center for Biomedical Informatics, Georgetown University Medical Center, Washington, DC
1.32. Department of Pathology and Immunology, Washington University in Saint Louis, Saint Louis, MO
1.33. Department of Medicine, Brown University, Providence, RI
1.34. Department of Biostatistics and Medical Informatics, University of Wisconsin-Madison, Madison, WI
1.35. Morgridge Institute for Research, Madison, WI
Deep learning describes a class of machine learning algorithms that are capable of combining raw inputs into layers of intermediate features. These algorithms have recently shown impressive results across a variety of domains. Biology and medicine are data-rich disciplines, but the data are complex and often ill-understood. Hence, deep learning techniques may be particularly well-suited to solve problems of these fields. We examine applications of deep learning to a variety of biomedical problems—patient classification, fundamental biological processes, and treatment of patients—and discuss whether deep learning will be able to transform these tasks or if the biomedical sphere poses unique challenges. Following from an extensive literature review, we find that deep learning has yet to revolutionize biomedicine or definitively resolve any of the most pressing challenges in the field, but promising advances have been made on the prior state of the art. Even though improvements over previous baselines have been modest in general, the recent progress indicates that deep learning methods will provide valuable means for speeding up or aiding human investigation. Though progress has been made linking a specific neural network’s prediction to input features, understanding how users should interpret these models to make testable hypotheses about the system under study remains an open challenge. Furthermore, the limited amount of labeled data for training presents problems in some domains, as do legal and privacy constraints on work with sensitive health records. Nonetheless, we foresee deep learning enabling changes at both bench and bedside with the potential to transform several areas of biology and medicine.
Biology and medicine are rapidly becoming data-intensive. A recent comparison of genomics with social media, online videos, and other data-intensive disciplines suggests that genomics alone will equal or surpass other fields in data generation and analysis within the next decade [1]. The volume and complexity of these data present new opportunities, but also pose new challenges. Automated algorithms that extract meaningful patterns could lead to actionable knowledge and change how we develop treatments, categorize patients, or study diseases, all within privacy-critical environments.
The term deep learning has come to refer to a collection of new techniques that, together, have demonstrated breakthrough gains over existing best-in-class machine learning algorithms across several fields. For example, over the past five years these methods have revolutionized image classification and speech recognition due to their flexibility and high accuracy [2]. More recently, deep learning algorithms have shown promise in fields as diverse as high-energy physics [3], computational chemistry [4], dermatology [5], and translation among written languages [6]. Across fields, “off-the-shelf” implementations of these algorithms have produced comparable or higher accuracy than previous best-in-class methods that required years of extensive customization, and specialized implementations are now being used at industrial scales.
Deep learning approaches grew from research on artificial neurons, which were first proposed in 1943 [7] as a model for how the neurons in a biological brain process information. The history of artificial neural networks—referred to as “neural networks” throughout this article—is interesting in its own right [8]. In neural networks, inputs are fed into the input layer, which feeds into one or more hidden layers, which eventually link to an output layer. A layer consists of a set of nodes, sometimes called “features” or “units,” which are connected via edges to the immediately earlier and the immediately deeper layers. In some special neural network architectures, nodes can connect to themselves with a delay. The nodes of the input layer generally consist of the variables being measured in the dataset of interest—for example, each node could represent the intensity value of a specific pixel in an image or the expression level of a gene in a specific transcriptomic experiment. The neural networks used for deep learning have multiple hidden layers. Each layer essentially performs feature construction for the layers before it. The training process used often allows layers deeper in the network to contribute to the refinement of earlier layers. For this reason, these algorithms can automatically engineer features that are suitable for many tasks and customize those features for one or more specific tasks.
Deep learning does many of the same things as more familiar machine learning approaches. In particular, deep learning approaches can be used both in supervised applications—where the goal is to accurately predict one or more labels or outcomes associated with each data point—in the place of regression approaches, as well as in unsupervised, or “exploratory” applications—where the goal is to summarize, explain, or identify interesting patterns in a data set—as a form of clustering. Deep learning methods may in fact combine both of these steps. When sufficient data are available and labeled, these methods construct features tuned to a specific problem and combine those features into a predictor. In fact, if the dataset is “labeled” with binary classes, a simple neural network with no hidden layers and no cycles between units is equivalent to logistic regression if the output layer is a sigmoid (logistic) function of the input layer. Similarly, for continuous outcomes, linear regression can be seen as a single-layer neural network. Thus, in some ways, supervised deep learning approaches can be seen as an extension of regression models that allow for greater flexibility and are especially well-suited for modeling non-linear relationships among the input features. Recently, hardware improvements and very large training datasets have allowed these deep learning techniques to surpass other machine learning algorithms for many problems. In a famous and early example, scientists from Google demonstrated that a neural network “discovered” that cats, faces, and pedestrians were important components of online videos [9] without being told to look for them. What if, more generally, deep learning takes advantage of the growth of data in biomedicine to tackle challenges in this field? Could these algorithms identify the “cats” hidden in our data—the patterns unknown to the researcher—and suggest ways to act on them? In this review, we examine deep learning’s application to biomedical science and discuss the unique challenges that biomedical data pose for deep learning methods.
Several important advances make the current surge of work done in this area possible. Easy-to-use software packages have brought the techniques of the field out of the specialist’s toolkit to a broad community of computational scientists. Additionally, new techniques for fast training have enabled their application to larger datasets [10]. Dropout of nodes, edges, and layers makes networks more robust, even when the number of parameters is very large. Finally, the larger datasets now available are also sufficient for fitting the many parameters that exist for deep neural networks. The convergence of these factors currently makes deep learning extremely adaptable and capable of addressing the nuanced differences of each domain to which it is applied.

This review discusses recent work in the biomedical domain, and most successful applications select neural network architectures that are well suited to the problem at hand. We sketch out a few simple example architectures in Figure 1. If data have a natural adjacency structure, a convolutional neural network (CNN) can take advantage of that structure by emphasizing local relationships, especially when convolutional layers are used in early layers of the neural network. Other neural network architectures such as autoencoders require no labels and are now regularly used for unsupervised tasks. In this review, we do not exhaustively discuss the different types of deep neural network architectures; an overview of the principal terms used herein is given in Table 1. Table 1 also provides select example applications, though in practice each neural network architecture has been broadly applied across multiple types of biomedical data. A recent book from Goodfellow et al. covers neural network architectures in detail [11], and LeCun et al. provide a more general introduction [2].
| Term | Definition | Example applications |
|---|---|---|
| Supervised learning | Machine-learning approaches with goal of prediction of labels or outcomes | |
| Unsupervised learning | Machine-learning approaches with goal of data summarization or pattern identification | |
| Neural network (NN) | Machine-learning approach inspired by biological neurons where inputs are fed into one or more layers, producing an output layer | |
| Deep neural network | NN with multiple hidden layers. Training happens over the network, and consequently such architectures allow for feature construction to occur alongside optimization of the overall training objective. | |
| Feed-forward neural network (FFNN) | NN that does not have cycles between nodes in the same layer | Most of the examples below are special cases of FFNNs, except recurrent neural networks. |
| Multi-layer perceptron (MLP) | Type of FFNN with at least one hidden layer where each deeper layer is a nonlinear function of each earlier layer | MLPs do not impose structure and are frequently used when there is no natural ordering of the inputs (e.g. as with gene expression measurements). |
| Convolutional neural network (CNN) | A NN with layers in which connectivity preserves local structure. If the data meet the underlying assumptions performance is often good, and such networks can require fewer examples to train effectively because they have fewer parameters and also provide improved efficiency. | CNNs are used for sequence data—such as DNA sequences—or grid data—such as medical and microscopy images. |
| Recurrent neural network (RNN) | A neural network with cycles between nodes within a hidden layer. | The RNN architecture is used for sequential data—such as clinical time series and text or genome sequences. |
| Long short-term memory (LSTM) neural network | This special type of RNN has features that enable models to capture longer-term dependencies. | LSTMs are gaining a substantial foothold in the analysis of natural language, and may become more widely applied to biological sequence data. |
| Autoencoder (AE) | A NN where the training objective is to minimize the error between the output layer and the input layer. Such neural networks are unsupervised and are often used for dimensionality reduction. | Autoencoders have been used for unsupervised analysis of gene expression data as well as data extracted from the electronic health record. |
| Variational autoencoder (VAE) | This special type of generative AE learns a probabilistic latent variable model. | VAEs have been shown to often produce meaningful reduced representations in the imaging domain, and some early publications have used VAEs to analyze gene expression data. |
| Denoising autoencoder (DA) | This special type of AE includes a step where noise is added to the input during the training process. The denoising step acts as smoothing and may allow for effective use on input data that is inherently noisy. | Like AEs, DAs have been used for unsupervised analysis of gene expression data as well as data extracted from the electronic health record. |
| Generative neural network | Neural networks that fall into this class can be used to generate data similar to input data. These models can be sampled to produce hypothetical examples. | A number of the unsupervised learning neural network architectures that are summarized here can be used in a generative fashion. |
| Restricted Boltzmann machine (RBM) | A generative NN that forms the building block for many deep learning approaches, having a single input layer and a single hidden layer, with no connections between the nodes within each layer | RBMs have been applied to combine multiple types of omic data (e.g. DNA methylation, mRNA expression, and miRNA expression). |
| Deep belief network (DBN) | Generative NN with several hidden layers, which can be obtained from combining multiple RBMs | DBNs can be used to predict new relationships in a drug-target interaction network. |
| Generative adversarial network (GAN) | A generative NN approach where two neural networks are trained. One neural network, the generator, is provided with a set of randomly generated inputs and tasked with generating samples. The second, the discriminator, is trained to differentiate real and generated samples. After the two neural networks are trained against each other, the resulting generator can be used to produce new examples. | GANs can synthesize new examples with the same statistical properties of datasets that contain individual-level records and are subject to sharing restrictions. They have also been applied to generate microscopy images. |
| Adversarial training | A process by which artificial training examples are maliciously designed to fool a NN and then input as training examples to make the resulting NN robust (no relation to GANs) | Adversarial training has been used in image analysis. |
| Data augmentation | A process by which transformations that do not affect relevant properties of the input data (e.g. arbitrary rotations of histopathology images) are applied to training examples to increase the size of the training set. | Data augmentation is widely used in the analysis of images because rotation transformations for biomedical images often do not change relevant properties of the image. |
While deep learning shows increased flexibility over other machine learning approaches, as seen in the remainder of this review, it requires large training sets in order to fit the hidden layers, as well as accurate labels for the supervised learning applications. For these reasons, deep learning has recently become popular in some areas of biology and medicine, while having lower adoption in other areas. At the same time, this highlights the potentially even larger role that it may play in future research, given the increases in data in all biomedical fields. It is also important to see it as a branch of machine learning and acknowledge that it has the same limitations as other approaches in that field. In particular, the results are still dependent on the underlying study design and the usual caveats of correlation versus causation still apply—a more precise answer is only better than a less precise one if it answers the correct question.
With this review, we ask the question: what is needed for deep learning to transform how we categorize, study, and treat individuals to maintain or restore health? We choose a high bar for “transform.” Andrew Grove, the former CEO of Intel, coined the term Strategic Inflection Point to refer to a change in technologies or environment that requires a business to be fundamentally reshaped [12]. Here, we seek to identify whether deep learning is an innovation that can induce a Strategic Inflection Point in the practice of biology or medicine.
There are already a number of reviews focused on applications of deep learning in biology [13,14,15,16,17], healthcare [18,19,20], and drug discovery [4,21,22,23]. Under our guiding question, we sought to highlight cases where deep learning enabled researchers to solve challenges that were previously considered infeasible or makes difficult, tedious analyses routine. We also identified approaches that researchers are using to sidestep challenges posed by biomedical data. We find that domain-specific considerations have greatly influenced how to best harness the power and flexibility of deep learning. Model interpretability is often critical. Understanding the patterns in data may be just as important as fitting the data. In addition, there are important and pressing questions about how to build networks that efficiently represent the underlying structure and logic of the data. Domain experts can play important roles in designing networks to represent data appropriately, encoding the most salient prior knowledge and assessing success or failure. There is also great potential to create deep learning systems that augment biologists and clinicians by prioritizing experiments or streamlining tasks that do not require expert judgment. We have divided the large range of topics into three broad classes: Disease and Patient Categorization, Fundamental Biological Study, and Treatment of Patients. Below, we briefly introduce the types of questions, approaches and data that are typical for each class in the application of deep learning.
A key challenge in biomedicine is the accurate classification of diseases and disease subtypes. In oncology, current “gold standard” approaches include histology, which requires interpretation by experts, or assessment of molecular markers such as cell surface receptors or gene expression. One example is the PAM50 approach to classifying breast cancer where the expression of 50 marker genes divides breast cancer patients into four subtypes. Substantial heterogeneity still remains within these four subtypes [24,25]. Given the increasing wealth of molecular data available, a more comprehensive subtyping seems possible. Several studies have used deep learning methods to better categorize breast cancer patients: For instance, denoising autoencoders, an unsupervised approach, can be used to cluster breast cancer patients [26], and CNNs can help count mitotic divisions, a feature that is highly correlated with disease outcome in histological images [27]. Despite these recent advances, a number of challenges exist in this area of research, most notably the integration of molecular and imaging data with other disparate types of data such as electronic health records (EHRs).
Deep learning can be applied to answer more fundamental biological questions; it is especially suited to leveraging large amounts of data from high-throughput “omics” studies. One classic biological problem where machine learning, and now deep learning, has been extensively applied is molecular target prediction. For example, deep recurrent neural networks (RNNs) have been used to predict gene targets of microRNAs [28], and CNNs have been applied to predict protein residue-residue contacts and secondary structure [29,30,31]. Other recent exciting applications of deep learning include recognition of functional genomic elements such as enhancers and promoters [32,33,34] and prediction of the deleterious effects of nucleotide polymorphisms [35].
Although the application of deep learning to patient treatment is just beginning, we expect new methods to recommend patient treatments, predict treatment outcomes, and guide the development of new therapies. One type of effort in this area aims to identify drug targets and interactions or predict drug response. Another uses deep learning on protein structures to predict drug interactions and drug bioactivity [36]. Drug repositioning using deep learning on transcriptomic data is another exciting area of research [37]. Restricted Boltzmann machines (RBMs) can be combined into deep belief networks (DBNs) to predict novel drug-target interactions and formulate drug repositioning hypotheses [38,39]. Finally, deep learning is also prioritizing chemicals in the early stages of drug discovery for new targets [23].
In healthcare, individuals are diagnosed with a disease or condition based on symptoms, the results of certain diagnostic tests, or other factors. Once diagnosed with a disease, an individual might be assigned a stage based on another set of human-defined rules. While these rules are refined over time, the process is evolutionary and ad hoc, potentially impeding the identification of underlying biological mechanisms and their corresponding treatment interventions.
Deep learning methods applied to a large corpus of patient phenotypes may provide a meaningful and more data-driven approach to patient categorization. For example, they may identify new shared mechanisms that would otherwise be obscured due to ad hoc historical definitions of disease. Perhaps deep neural networks, by reevaluating data without the context of our assumptions, can reveal novel classes of treatable conditions.
In spite of such optimism, the ability of deep learning models to indiscriminately extract predictive signals must also be assessed and operationalized with care. Imagine a deep neural network is provided with clinical test results gleaned from electronic health records. Because physicians may order certain tests based on their suspected diagnosis, a deep neural network may learn to “diagnose” patients simply based on the tests that are ordered. For some objective functions, such as predicting an International Classification of Diseases (ICD) code, this may offer good performance even though it does not provide insight into the underlying disease beyond physician activity. This challenge is not unique to deep learning approaches; however, it is important for practitioners to be aware of these challenges and the possibility in this domain of constructing highly predictive classifiers of questionable utility.
Our goal in this section is to assess the extent to which deep learning is already contributing to the discovery of novel categories. Where it is not, we focus on barriers to achieving these goals. We also highlight approaches that researchers are taking to address challenges within the field, particularly with regards to data availability and labeling.
Deep learning methods have transformed the analysis of natural images and video, and similar examples are beginning to emerge with medical images. Deep learning has been used to classify lesions and nodules; localize organs, regions, landmarks and lesions; segment organs, organ substructures and lesions; retrieve images based on content; generate and enhance images; and combine images with clinical reports [19,40].
Though there are many commonalities with the analysis of natural images, there are also key differences. In all cases that we examined, fewer than one million images were available for training, and datasets are often many orders of magnitude smaller than collections of natural images. Researchers have developed subtask-specific strategies to address this challenge.
Data augmentation provides an effective strategy for working with small training sets. The practice is exemplified by a series of papers that analyze images from mammographies [41,42,43,44,45]. To expand the number and diversity of images, researchers constructed adversarial [44] or augmented [45] examples. Adversarial training examples are constructed by selecting targeted small transformations to input data that cause a model to produce very different outputs. Augmented training applies perturbations to the input data that do not change the underlying meaning, such as rotations for pathology images. An alternative in the domain is to train towards human-created features before subsequent fine-tuning [42], which can help to sidestep this challenge though it does give up deep learning techniques’ strength as feature constructors.
A second strategy repurposes features extracted from natural images by deep learning models, such as ImageNet [46], for new purposes. Diagnosing diabetic retinopathy through color fundus images became an area of focus for deep learning researchers after a large labeled image set was made publicly available during a 2015 Kaggle competition [47]. Most participants trained neural networks from scratch [47,48,49], but Gulshan et al. [50] repurposed a 48-layer Inception-v3 deep architecture pre-trained on natural images and surpassed the state-of-the-art specificity and sensitivity. Such features were also repurposed to detect melanoma, the deadliest form of skin cancer, from dermoscopic [51,52] and non-dermoscopic images of skin lesions [5,53,54] as well as age-related macular degeneration [55]. Pre-training on natural images can enable very deep networks to succeed without overfitting. For the melanoma task, reported performance was competitive with or better than a board of certified dermatologists [5,51]. Reusing features from natural images is also an emerging approach for radiographic images, where datasets are often too small to train large deep neural networks without these techniques [56,57,58,59]. A deep CNN trained on natural images boosts performance in radiographic images [58]. However, the target task required either re-training the initial model from scratch with special pre-processing or fine-tuning of the whole network on radiographs with heavy data augmentation to avoid overfitting.
The technique of reusing features from a different task falls into the broader area of transfer learning (see Discussion). Though we’ve mentioned numerous successes for the transfer of natural image features to new tasks, we expect that a lower proportion of negative results have been published. The analysis of magnetic resonance images (MRIs) is also faced with the challenge of small training sets. In this domain, Amit et al. [60] investigated the tradeoff between pre-trained models from a different domain and a small CNN trained only with MRI images. In contrast with the other selected literature, they found a smaller network trained with data augmentation on a few hundred images from a few dozen patients can outperform a pre-trained out-of-domain classifier.
Another way of dealing with limited training data is to divide rich data—e.g. 3D images—into numerous reduced projections. Shin et al. [57] compared various deep network architectures, dataset characteristics, and training procedures for computer tomography-based (CT) abnormality detection. They concluded that networks as deep as 22 layers could be useful for 3D data, despite the limited size of training datasets. However, they noted that choice of architecture, parameter setting, and model fine-tuning needed is very problem- and dataset-specific. Moreover, this type of task often depends on both lesion localization and appearance, which poses challenges for CNN-based approaches. Straightforward attempts to capture useful information from full-size images in all three dimensions simultaneously via standard neural network architectures were computationally unfeasible. Instead, two-dimensional models were used to either process image slices individually (2D) or aggregate information from a number of 2D projections in the native space (2.5D).
Roth et al. compared 2D, 2.5D, and 3D CNNs on a number of tasks for computer-aided detection from CT scans and showed that 2.5D CNNs performed comparably well to 3D analogs, while requiring much less training time, especially on augmented training sets [61]. Another advantage of 2D and 2.5D networks is the wider availability of pre-trained models. However, reducing the dimensionality is not always helpful. Nie et al. [62] showed that multimodal, multi-channel 3D deep architecture was successful at learning high-level brain tumor appearance features jointly from MRI, functional MRI, and diffusion MRI images, outperforming single-modality or 2D models. Overall, the variety of modalities, properties and sizes of training sets, the dimensionality of input, and the importance of end goals in medical image analysis are provoking a development of specialized deep neural network architectures, training and validation protocols, and input representations that are not characteristic of widely-studied natural images.
Predictions from deep neural networks can be evaluated for use in workflows that also incorporate human experts. In a large dataset of mammography images, Kooi et al. [63] demonstrated that deep neural networks outperform a traditional computer-aided diagnosis system at low sensitivity and perform comparably at high sensitivity. They also compared network performance to certified screening radiologists on a patch level and found no significant difference between the network and the readers. However, using deep methods for clinical practice is challenged by the difficulty of assigning a level of confidence to each prediction. Leibig et al. [49] estimated the uncertainty of deep networks for diabetic retinopathy diagnosis by linking dropout networks with approximate Bayesian inference. Techniques that assign confidences to each prediction should aid physician-computer interactions and improve uptake by physicians.
Systems to aid in the analysis of histology slides are also promising use cases for deep learning [64]. Ciresan et al. [27] developed one of the earliest approaches for histology slides, winning the 2012 International Conference on Pattern Recognition’s Contest on Mitosis Detection while achieving human-competitive accuracy. In more recent work, Wang et al. [65] analyzed stained slides of lymph node slices to identify cancers. On this task a pathologist has about a 3% error rate. The pathologist did not produce any false positives, but did have a number of false negatives. The algorithm had about twice the error rate of a pathologist, but the errors were not strongly correlated. Combining pre-trained deep network architectures with multiple augmentation techniques enabled accurate detection of breast cancer from a very small set of histology images with less than 100 images per class [66]. In this area, these algorithms may be ready to be incorporated into existing tools to aid pathologists and reduce the false negative rate. Ensembles of deep learning and human experts may help overcome some of the challenges presented by data limitations.
One source of training examples with rich phenotypical annotations is the EHR. Billing information in the form of ICD codes are simple annotations but phenotypic algorithms can combine laboratory tests, medication prescriptions, and patient notes to generate more reliable phenotypes. Recently, Lee et al. [67] developed an approach to distinguish individuals with age-related macular degeneration from control individuals. They trained a deep neural network on approximately 100,000 images extracted from structured electronic health records, reaching greater than 93% accuracy. The authors used their test set to evaluate when to stop training. In other domains, this has resulted in a minimal change in the estimated accuracy [68], but we recommend the use of an independent test set whenever feasible.
Rich clinical information is stored in EHRs. However, manually annotating a large set requires experts and is time consuming. For chest X-ray studies, a radiologist usually spends a few minutes per example. Generating the number of examples needed for deep learning is infeasibly expensive. Instead, researchers may benefit from using text mining to generate annotations [69], even if those annotations are of modest accuracy. Wang et al. [70] proposed to build predictive deep neural network models through the use of images with weak labels. Such labels are automatically generated and not verified by humans, so they may be noisy or incomplete. In this case, they applied a series of natural language processing (NLP) techniques to the associated chest X-ray radiological reports. They first extracted all diseases mentioned in the reports using a state-of-the-art NLP tool, then applied a new method, NegBio [71], to filter negative and equivocal findings in the reports. Evaluation on four independent datasets demonstrated that NegBio is highly accurate for detecting negative and equivocal findings (~90% in F₁ score, which balances precision and recall [72]). The resulting dataset [73] consisted of 112,120 frontal-view chest X-ray images from 30,805 patients, and each image was associated with one or more text-mined (weakly-labeled) pathology categories (e.g. pneumonia and cardiomegaly) or “no finding” otherwise. Further, Wang et al. [70] used this dataset with a unified weakly-supervised multi-label image classification framework to detect common thoracic diseases. It showed superior performance over a benchmark using fully-labeled data.
Another example of semi-automated label generation for hand radiograph segmentation employed positive mining, an iterative procedure that combines manual labeling with automatic processing [74]. First, the initial training set was created by manually labeling 100 of 12,600 unlabeled radiographs that were used to train a model and predict labels for the rest of the dataset. Then, poor quality predictions were discarded through manual inspection, the initial training set was expanded with the acceptable segmentations, and the process was repeated. This procedure had to be repeated six times to obtain good quality segmentation labeling for all radiographs, except for 100 corner cases that still required manual annotation. These annotations allowed accurate segmentation of all hand images in the test set and boosted the final performance in radiograph classification [74].
With the exception of natural image-like problems (e.g. melanoma detection), biomedical imaging poses a number of challenges for deep learning. Datasets are typically small, annotations can be sparse, and images are often high-dimensional, multimodal, and multi-channel. Techniques like transfer learning, heavy dataset augmentation, and the use of multi-view and multi-stream architectures are more common than in the natural image domain. Furthermore, high model sensitivity and specificity can translate directly into clinical value. Thus, prediction evaluation, uncertainty estimation, and model interpretation methods are also of great importance in this domain (see Discussion). Finally, there is a need for better pathologist-computer interaction techniques that will allow combining the power of deep learning methods with human expertise and lead to better-informed decisions for patient treatment and care.
Due to the rapid growth of scholarly publications and EHRs, biomedical text mining has become increasingly important in recent years. The main tasks in biological and clinical text mining include, but are not limited to, named entity recognition, relation/event extraction, and information retrieval (Figure 2). Deep learning is appealing in this domain because of its competitive performance versus traditional methods and ability to overcome challenges in feature engineering. Relevant applications can be stratified by the application domain (biomedical literature vs. clinical notes) and the actual task (e.g. concept or relation extraction).
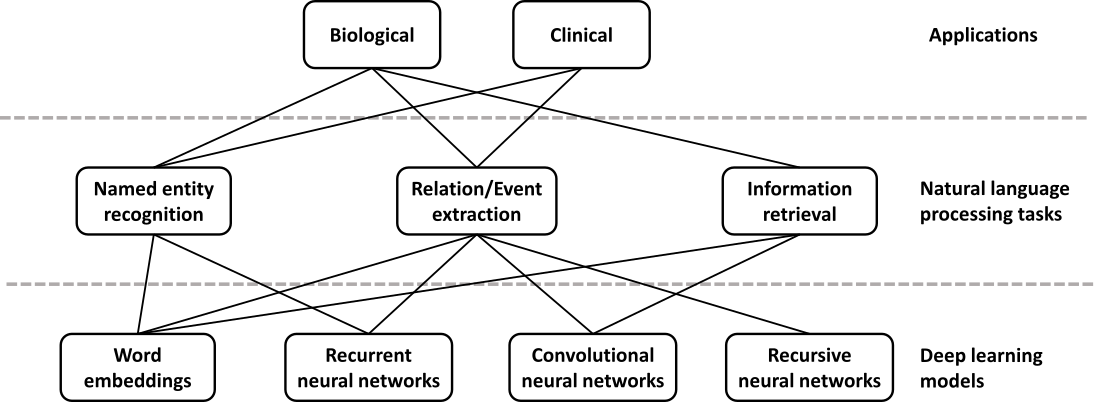
Named entity recognition (NER) is a task of identifying text spans that refer to a biological concept of a specific class, such as disease or chemical, in a controlled vocabulary or ontology. NER is often needed as a first step in many complex text mining systems. The current state-of-the-art methods typically reformulate the task as a sequence labeling problem and use conditional random fields [75,76,77]. In recent years, word embeddings that contain rich latent semantic information of words have been widely used to improve the NER performance. Liu et al. studied the effect of word embeddings on drug name recognition and compared them with traditional semantic features [78]. Tang et al. investigated word embeddings in gene, DNA, and cell line mention detection tasks [79]. Moreover, Wu et al. examined the use of neural word embeddings for clinical abbreviation disambiguation [80]. Liu et al. exploited task-oriented resources to learn word embeddings for clinical abbreviation expansion [81].
Relation extraction involves detecting and classifying semantic relationships between entities from the literature. At present, kernel methods or feature-based approaches are commonly applied [82,83,84]. Deep learning can relieve the feature sparsity and engineering problems. Some studies focused on jointly extracting biomedical entities and relations simultaneously [85,86], while others applied deep learning on relation classification given the relevant entities. For example, both multichannel dependency-based CNNs [87] and shortest path-based CNNs [88,89] are well-suited for sentence-based protein-protein extraction. Jiang et al. proposed a biomedical domain-specific word embedding model to reduce the manual labor of designing semantic representation for the same task [90]. Gu et al. employed a maximum entropy model and a CNN model for chemical-induced disease relation extraction at the inter- and intra-sentence level, respectively [91]. For drug-drug interactions, Zhao et al. used a CNN that employs word embeddings with the syntactic information of a sentence as well as features of part-of-speech tags and dependency trees [92]. Asada et al. experimented with an attention CNN [93], and Yi et al. proposed an RNN model with multiple attention layers [94]. In both cases, it is a single model with attention mechanism, which allows the decoder to focus on different parts of the source sentence. As a result, it does not require dependency parsing or training multiple models. Both attention CNN and RNN have comparable results, but the CNN model has an advantage in that it can be easily computed in parallel, hence making it faster with recent graphics processing units (GPUs).
For biotopes event extraction, Li et al. employed CNNs and distributed representation [95] while Mehryary et al. used long short-term memory (LSTM) networks to extract complicated relations [96]. Li et al. applied word embedding to extract complete events from biomedical text and achieved results comparable to the state-of-the-art systems [97]. There are also approaches that identify event triggers rather than the complete event [98,99]. Taken together, deep learning models outperform traditional kernel methods or feature-based approaches by 1–5% in f-score. Among various deep learning approaches, CNNs stand out as the most popular model both in terms of computational complexity and performance, while RNNs have achieved continuous progress.
Information retrieval is a task of finding relevant text that satisfies an information need from within a large document collection. While deep learning has not yet achieved the same level of success in this area as seen in others, the recent surge of interest and work suggest that this may be quickly changing. For example, Mohan et al. described a deep learning approach to modeling the relevance of a document’s text to a query, which they applied to the entire biomedical literature [100].
To summarize, deep learning has shown promising results in many biomedical text mining tasks and applications. However, to realize its full potential in this domain, either large amounts of labeled data or technical advancements in current methods coping with limited labeled data are required.
EHR data include substantial amounts of free text, which remains challenging to approach [101]. Often, researchers developing algorithms that perform well on specific tasks must design and implement domain-specific features [102]. These features capture unique aspects of the literature being processed. Deep learning methods are natural feature constructors. In recent work, Chalapathy et al. evaluated the extent to which deep learning methods could be applied on top of generic features for domain-specific concept extraction [103]. They found that performance was in line with, but lower than the best domain-specific method [103]. This raises the possibility that deep learning may impact the field by reducing the researcher time and cost required to develop specific solutions, but it may not always lead to performance increases.
In recent work, Yoon et al. [104] analyzed simple features using deep neural networks and found that the patterns recognized by the algorithms could be re-used across tasks. Their aim was to analyze the free text portions of pathology reports to identify the primary site and laterality of tumors. The only features the authors supplied to the algorithms were unigrams (counts for single words) and bigrams (counts for two-word combinations) in a free text document. They subset the full set of words and word combinations to the 400 most common. The machine learning algorithms that they employed (naïve Bayes, logistic regression, and deep neural networks) all performed relatively similarly on the task of identifying the primary site. However, when the authors evaluated the more challenging task, evaluating the laterality of each tumor, the deep neural network outperformed the other methods. Of particular interest, when the authors first trained a neural network to predict the primary site and then repurposed those features as a component of a secondary neural network trained to predict laterality, the performance was higher than a laterality-trained neural network. This demonstrates how deep learning methods can repurpose features across tasks, improving overall predictions as the field tackles new challenges. The Discussion further reviews this type of transfer learning.
Several authors have created reusable feature sets for medical terminologies using natural language processing and neural embedding models, as popularized by word2vec [105]. Minarro-Giménez et al. [106] applied the word2vec deep learning toolkit to medical corpora and evaluated the efficiency of word2vec in identifying properties of pharmaceuticals based on mid-sized, unstructured medical text corpora without any additional background knowledge. A goal of learning terminologies for different entities in the same vector space is to find relationships between different domains (e.g. drugs and the diseases they treat). It is difficult for us to provide a strong statement on the broad utility of these methods. Manuscripts in this area tend to compare algorithms applied to the same data but lack a comparison against overall best-practices for one or more tasks addressed by these methods. Techniques have been developed for free text medical notes [107], ICD and National Drug Codes [108,109], and claims data [110]. Methods for neural embeddings learned from electronic health records have at least some ability to predict disease-disease associations and implicate genes with a statistical association with a disease [111], but the evaluations performed did not differentiate between simple predictions (i.e. the same disease in different sites of the body) and non-intuitive ones. Jagannatha and Yu [112] further employed a bidirectional LSTM structure to extract adverse drug events from electronic health records, and Lin et al. [113] investigated using CNNs to extract temporal relations. While promising, a lack of rigorous evaluation of the real-world utility of these kinds of features makes current contributions in this area difficult to evaluate. Comparisons need to be performed to examine the true utility against leading approaches (i.e. algorithms and data) as opposed to simply evaluating multiple algorithms on the same potentially limited dataset.
Identifying consistent subgroups of individuals and individual health trajectories from clinical tests is also an active area of research. Approaches inspired by deep learning have been used for both unsupervised feature construction and supervised prediction. Early work by Lasko et al. [114], combined sparse autoencoders and Gaussian processes to distinguish gout from leukemia from uric acid sequences. Later work showed that unsupervised feature construction of many features via denoising autoencoder neural networks could dramatically reduce the number of labeled examples required for subsequent supervised analyses [115]. In addition, it pointed towards features learned during unsupervised training being useful for visualizing and stratifying subgroups of patients within a single disease. In a concurrent large-scale analysis of EHR data from 700,000 patients, Miotto et al. [116] used a deep denoising autoencoder architecture applied to the number and co-occurrence of clinical events to learn a representation of patients (DeepPatient). The model was able to predict disease trajectories within one year with over 90% accuracy, and patient-level predictions were improved by up to 15% when compared to other methods. Choi et al. [117] attempted to model the longitudinal structure of EHRs with an RNN to predict future diagnosis and medication prescriptions on a cohort of 260,000 patients followed for 8 years (Doctor AI). Pham et al. [118] built upon this concept by using an RNN with a LSTM architecture enabling explicit modelling of patient trajectories through the use of memory cells. The method, DeepCare, performed better than shallow models or plain RNN when tested on two independent cohorts for its ability to predict disease progression, intervention recommendation and future risk prediction. Nguyen et al. [119] took a different approach and used word embeddings from EHRs to train a CNN that could detect and pool local clinical motifs to predict unplanned readmission after six months, with performance better than the baseline method (Deepr). Razavian et al. [120] used a set of 18 common lab tests to predict disease onset using both CNN and LSTM architectures and demonstrated an improvement over baseline regression models. However, numerous challenges including data integration (patient demographics, family history, laboratory tests, text-based patient records, image analysis, genomic data) and better handling of streaming temporal data with many features will need to be overcome before we can fully assess the potential of deep learning for this application area.
Still, recent work has also revealed domains in which deep networks have proven superior to traditional methods. Survival analysis models the time leading to an event of interest from a shared starting point, and in the context of EHR data, often associates these events to subject covariates. Exploring this relationship is difficult, however, given that EHR data types are often heterogeneous, covariates are often missing, and conventional approaches require the covariate-event relationship be linear and aligned to a specific starting point [121]. Early approaches, such as the Faraggi-Simon feed-forward network, aimed to relax the linearity assumption, but performance gains were lacking [122]. Katzman et al. in turn developed a deep implementation of the Faraggi-Simon network that, in addition to outperforming Cox regression, was capable of comparing the risk between a given pair of treatments, thus potentially acting as recommender system [123]. To overcome the remaining difficulties, researchers have turned to deep exponential families, a class of latent generative models that are constructed from any type of exponential family distributions [124]. The result was a deep survival analysis model capable of overcoming challenges posed by missing data and heterogeneous data types, while uncovering nonlinear relationships between covariates and failure time. They showed their model more accurately stratified patients as a function of disease risk score compared to the current clinical implementation.
There is a computational cost for these methods, however, when compared to traditional, non-neural network approaches. For the exponential family models, despite their scalability [125], an important question for the investigator is whether he or she is interested in estimates of posterior uncertainty. Given that these models are effectively Bayesian neural networks, much of their utility simplifies to whether a Bayesian approach is warranted for a given increase in computational cost. Moreover, as with all variational methods, future work must continue to explore just how well the posterior distributions are approximated, especially as model complexity increases [126].
A dearth of true labels is perhaps among the biggest obstacles for EHR-based analyses that employ machine learning. Popular deep learning (and other machine learning) methods are often used to tackle classification tasks and thus require ground-truth labels for training. For EHRs this can mean that researchers must hire multiple clinicians to manually read and annotate individual patients’ records through a process called chart review. This allows researchers to assign “true” labels, i.e. those that match our best available knowledge. Depending on the application, sometimes the features constructed by algorithms also need to be manually validated and interpreted by clinicians. This can be time consuming and expensive [127]. Because of these costs, much of this research, including the work cited in this review, skips the process of expert review. Clinicians’ skepticism for research without expert review may greatly dampen their enthusiasm for the work and consequently reduce its impact. To date, even well-resourced large national consortia have been challenged by the task of acquiring enough expert-validated labeled data. For instance, in the eMERGE consortia and PheKB database [128], most samples with expert validation contain only 100 to 300 patients. These datasets are quite small even for simple machine learning algorithms. The challenge is greater for deep learning models with many parameters. While unsupervised and semi-supervised approaches can help with small sample sizes, the field would benefit greatly from large collections of anonymized records in which a substantial number of records have undergone expert review. This challenge is not unique to EHR-based studies. Work on medical images, omics data in applications for which detailed metadata are required, and other applications for which labels are costly to obtain will be hampered as long as abundant curated data are unavailable.
Successful approaches to date in this domain have sidestepped this challenge by making methodological choices that either reduce the need for labeled examples or that use transformations to training data to increase the number of times it can be used before overfitting occurs. For example, the unsupervised and semi-supervised methods that we have discussed reduce the need for labeled examples [115]. The anchor and learn framework [129] uses expert knowledge to identify high-confidence observations from which labels can be inferred. If transformations are available that preserve the meaningful content of the data, the adversarial and augmented training techniques discussed above can reduce overfitting. While these can be easily imagined for certain methods that operate on images, it is more challenging to figure out equivalent transformations for a patient’s clinical test results. Consequently, it may be hard to employ such training examples with other applications. Finally, approaches that transfer features can also help use valuable training data most efficiently. Rajkomar et al. trained a deep neural network using generic images before tuning using only radiology images [58]. Datasets that require many of the same types of features might be used for initial training, before fine tuning takes place with the more sparse biomedical examples. Though the analysis has not yet been attempted, it is possible that analogous strategies may be possible with electronic health records. For example, features learned from the electronic health record for one type of clinical test (e.g. a decrease over time in a lab value) may transfer across phenotypes. Methods to accomplish more with little high-quality labeled data arose in other domains and may also be adapted to this challenge, e.g. data programming [130]. In data programming, noisy automated labeling functions are integrated.
Numerous commentators have described data as the new oil [131,132]. The idea behind this metaphor is that data are available in large quantities, valuable once refined, and this underlying resource will enable a data-driven revolution in how work is done. Contrasting with this perspective, Ratner, Bach, and Ré described labeled training data, instead of data, as “The New New Oil” [133]. In this framing, data are abundant and not a scarce resource. Instead, new approaches to solving problems arise when labeled training data become sufficient to enable them. Based on our review of research on deep learning methods to categorize disease, the latter framing rings true.
We expect improved methods for domains with limited data to play an important role if deep learning is going to transform how we categorize states of human health. We don’t expect that deep learning methods will replace expert review. We expect them to complement expert review by allowing more efficient use of the costly practice of manual annotation.
To construct the types of very large datasets that deep learning methods thrive on, we need robust sharing of large collections of data. This is in part a cultural challenge. We touch on this challenge in the Discussion section. Beyond the cultural hurdles around data sharing, there are also technological and legal hurdles related to sharing individual health records or deep models built from such records. This subsection deals primarily with these challenges.
EHRs are designed chiefly for clinical, administrative and financial purposes, such as patient care, insurance, and billing [134]. Science is at best a tertiary priority, presenting challenges to EHR-based research in general and to deep learning research in particular. Although there is significant work in the literature around EHR data quality and the impact on research [135], we focus on three types of challenges: local bias, wider standards, and legal issues. Note these problems are not restricted to EHRs but can also apply to any large biomedical dataset, e.g. clinical trial data.
Even within the same healthcare system, EHRs can be used differently [136,137]. Individual users have unique documentation and ordering patterns, with different departments and different hospitals having different priorities that code patients and introduce missing data in a non-random fashion [138]. Patient data may be kept across several “silos” within a single health system (e.g. separate nursing documentation, registries, etc.). Even the most basic task of matching patients across systems can be challenging due to data entry issues [139]. The situation is further exacerbated by the ongoing introduction, evolution, and migration of EHR systems, especially where reorganized and acquired healthcare facilities have to merge. Further, even the ostensibly least-biased data type, laboratory measurements, can be biased based by both the healthcare process and patient health state [140]. As a result, EHR data can be less complete and less objective than expected.
In the wider picture, standards for EHRs are numerous and evolving. Proprietary systems, indifferent and scattered use of health information standards, and controlled terminologies makes combining and comparison of data across systems challenging [141]. Further diversity arises from variation in languages, healthcare practices, and demographics. Merging EHRs gathered in different systems (and even under different assumptions) is challenging [142].
Combining or replicating studies across systems thus requires controlling for both the above biases and dealing with mismatching standards. This has the practical effect of reducing cohort size, limiting statistical significance, preventing the detection of weak effects [143], and restricting the number of parameters that can be trained in a model. Further, rule-based algorithms have been popular in EHR-based research, but because these are developed at a single institution and trained with a specific patient population, they do not transfer easily to other healthcare systems [144]. Genetic studies using EHR data are subject to even more bias, as the differences in population ancestry across health centers (e.g. proportion of patients with African or Asian ancestry) can affect algorithm performance. For example, Wiley et al. [145] showed that warfarin dosing algorithms often under-perform in African Americans, illustrating that some of these issues are unresolved even at a treatment best practices level. Lack of standardization also makes it challenging for investigators skilled in deep learning to enter the field, as numerous data processing steps must be performed before algorithms are applied.
Finally, even if data were perfectly consistent and compatible across systems, attempts to share and combine EHR data face considerable legal and ethical barriers. Patient privacy can severely restrict the sharing and use of EHR data [146]. Here again, standards are heterogeneous and evolving, but often EHR data cannot be exported or even accessed directly for research purposes without appropriate consent. In the United States, research use of EHR data is subject both to the Common Rule and the Health Insurance Portability and Accountability Act (HIPAA). Ambiguity in the regulatory language and individual interpretation of these rules can hamper use of EHR data [147]. Once again, this has the effect of making data gathering more laborious and expensive, reducing sample size and study power.
Several technological solutions have been proposed in this direction, allowing access to sensitive data satisfying privacy and legal concerns. Software like DataShield [148] and ViPAR [149], although not EHR-specific, allow querying and combining of datasets and calculation of summary statistics across remote sites by “taking the analysis to the data”. The computation is carried out at the remote site. Conversely, the EH4CR project [141] allows analysis of private data by use of an inter-mediation layer that interprets remote queries across internal formats and datastores and returns the results in a de-identified standard form, thus giving real-time consistent but secure access. Continuous Analysis [150] can allow reproducible computing on private data. Using such techniques, intermediate results can be automatically tracked and shared without sharing the original data. While none of these have been used in deep learning, the potential is there.
Even without sharing data, algorithms trained on confidential patient data may present security risks or accidentally allow for the exposure of individual level patient data. Tramer et al. [151] showed the ability to steal trained models via public application programming interfaces (APIs). Dwork and Roth [152] demonstrate the ability to expose individual level information from accurate answers in a machine learning model. Attackers can use similar attacks to find out if a particular data instance was present in the original training set for the machine learning model [153], in this case, whether a person’s record was present. To protect against these attacks, Simmons et al. [154] developed the ability to perform genome-wide association studies (GWASs) in a differentially private manner, and Abadi et al. [155] show the ability to train deep learning classifiers under the differential privacy framework.
These attacks also present a potential hazard for approaches that aim to generate data. Choi et al. propose generative adversarial neural networks (GANs) as a tool to make sharable EHR data [156], and Esteban et al. [157] showed that recurrent GANs could be used for time series data. However, in both cases the authors did not take steps to protect the model from such attacks. There are approaches to protect models, but they pose their own challenges. Training in a differentially private manner provides a limited guarantee that an algorithm’s output will be equally likely to occur regardless of the participation of any one individual. The limit is determined by parameters which provide a quantification of privacy. Beaulieu-Jones et al. demonstrated the ability to generate data that preserved properties of the SPRINT clinical trial with GANs under the differential privacy framework [158]. Both Beaulieu-Jones et al. and Esteban et al. train models on synthetic data generated under differential privacy and observe performance from a transfer learning evaluation that is only slightly below models trained on the original, real data. Taken together, these results suggest that differentially private GANs may be an attractive way to generate sharable datasets for downstream reanalysis.
Federated learning [159] and secure aggregations [160,161] are complementary approaches that reinforce differential privacy. Both aim to maintain privacy by training deep learning models from decentralized data sources such as personal mobile devices without transferring actual training instances. This is becoming of increasing importance with the rapid growth of mobile health applications. However, the training process in these approaches places constraints on the algorithms used and can make fitting a model substantially more challenging. It can be trivial to train a model without differential privacy, but quite difficult to train one within the differential privacy framework [158]. This problem can be particularly pronounced with small sample sizes.
While none of these problems are insurmountable or restricted to deep learning, they present challenges that cannot be ignored. Technical evolution in EHRs and data standards will doubtless ease—although not solve—the problems of data sharing and merging. More problematic are the privacy issues. Those applying deep learning to the domain should consider the potential of inadvertently disclosing the participants’ identities. Techniques that enable training on data without sharing the raw data may have a part to play. Training within a differential privacy framework may often be warranted.
In April 2016, the European Union adopted new rules regarding the use of personal information, the General Data Protection Regulation [162]. A component of these rules can be summed up by the phrase “right to an explanation”. Those who use machine learning algorithms must be able to explain how a decision was reached. For example, a clinician treating a patient who is aided by a machine learning algorithm may be expected to explain decisions that use the patient’s data. The new rules were designed to target categorization or recommendation systems, which inherently profile individuals. Such systems can do so in ways that are discriminatory and unlawful.
As datasets become larger and more complex, we may begin to identify relationships in data that are important for human health but difficult to understand. The algorithms described in this review and others like them may become highly accurate and useful for various purposes, including within medical practice. However, to discover and avoid discriminatory applications it will be important to consider interpretability alongside accuracy. A number of properties of genomic and healthcare data will make this difficult.
First, research samples are frequently non-representative of the general population of interest; they tend to be disproportionately sick [163], male [164], and European in ancestry [165]. One well-known consequence of these biases in genomics is that penetrance is consistently lower in the general population than would be implied by case-control data, as reviewed in [163]. Moreover, real genetic associations found in one population may not hold in other populations with different patterns of linkage disequilibrium (even when population stratification is explicitly controlled for [166]). As a result, many genomic findings are of limited value for people of non-European ancestry [165] and may even lead to worse treatment outcomes for them. Methods have been developed for mitigating some of these problems in genomic studies [163,166], but it is not clear how easily they can be adapted for deep models that are designed specifically to extract subtle effects from high-dimensional data. For example, differences in the equipment that tended to be used for cases versus controls have led to spurious genetic findings (e.g. Sebastiani et al.’s retraction [167]). In some contexts, it may not be possible to correct for all of these differences to the degree that a deep network is unable to use them. Moreover, the complexity of deep networks makes it difficult to determine when their predictions are likely to be based on such nominally-irrelevant features of the data (called “leakage” in other fields [168]). When we are not careful with our data and models, we may inadvertently say more about the way the data was collected (which may involve a history of unequal access and discrimination) than about anything of scientific or predictive value. This fact can undermine the privacy of patient data [168] or lead to severe discriminatory consequences [169].
There is a small but growing literature on the prevention and mitigation of data leakage [168], as well as a closely-related literature on discriminatory model behavior [170], but it remains difficult to predict when these problems will arise, how to diagnose them, and how to resolve them in practice. There is even disagreement about which kinds of algorithmic outcomes should be considered discriminatory [171]. Despite the difficulties and uncertainties, machine learning practitioners (and particularly those who use deep neural networks, which are challenging to interpret) must remain cognizant of these dangers and make every effort to prevent harm from discriminatory predictions. To reach their potential in this domain, deep learning methods will need to be interpretable (see Discussion). Researchers need to consider the extent to which biases may be learned by the model and whether or not a model is sufficiently interpretable to identify bias. We discuss the challenge of model interpretability more thoroughly in Discussion.
Longitudinal analysis follows a population across time, for example, prospectively from birth or from the onset of particular conditions. In large patient populations, longitudinal analyses such as the Framingham Heart Study [172] and the Avon Longitudinal Study of Parents and Children [173] have yielded important discoveries about the development of disease and the factors contributing to health status. Yet, a common practice in EHR-based research is to take a snapshot at a point in time and convert patient data to a traditional vector for machine learning and statistical analysis. This results in loss of information as timing and order of events can provide insight into a patient’s disease and treatment [174]. Efforts to model sequences of events have shown promise [175] but require exceedingly large patient sizes due to discrete combinatorial bucketing. Lasko et al. [114] used autoencoders on longitudinal sequences of serum uric acid measurements to identify population subtypes. More recently, deep learning has shown promise working with both sequences (CNNs) [176] and the incorporation of past and current state (RNNs, LSTMs) [118]. This may be a particular area of opportunity for deep neural networks. The ability to recognize relevant sequences of events from a large number of trajectories requires powerful and flexible feature construction methods—an area in which deep neural networks excel.
The study of cellular structure and core biological processes—transcription, translation, signaling, metabolism, etc.—in humans and model organisms will greatly impact our understanding of human disease over the long horizon [177]. Predicting how cellular systems respond to environmental perturbations and are altered by genetic variation remain daunting tasks. Deep learning offers new approaches for modeling biological processes and integrating multiple types of omic data [178], which could eventually help predict how these processes are disrupted in disease. Recent work has already advanced our ability to identify and interpret genetic variants, study microbial communities, and predict protein structures, which also relates to the problems discussed in the drug development section. In addition, unsupervised deep learning has enormous potential for discovering novel cellular states from gene expression, fluorescence microscopy, and other types of data that may ultimately prove to be clinically relevant.
Progress has been rapid in genomics and imaging, fields where important tasks are readily adapted to well-established deep learning paradigms. One-dimensional convolutional and recurrent neural networks are well-suited for tasks related to DNA- and RNA-binding proteins, epigenomics, and RNA splicing. Two dimensional CNNs are ideal for segmentation, feature extraction, and classification in fluorescence microscopy images [17]. Other areas, such as cellular signaling, are biologically important but studied less-frequently to date, with some exceptions [179]. This may be a consequence of data limitations or greater challenges in adapting neural network architectures to the available data. Here, we highlight several areas of investigation and assess how deep learning might move these fields forward.
Gene expression technologies characterize the abundance of many thousands of RNA transcripts within a given organism, tissue, or cell. This characterization can represent the underlying state of the given system and can be used to study heterogeneity across samples as well as how the system reacts to perturbation. While gene expression measurements were traditionally made by quantitative polymerase chain reaction (qPCR), low-throughput fluorescence-based methods, and microarray technologies, the field has shifted in recent years to primarily performing RNA sequencing (RNA-seq) to catalog whole transcriptomes. As RNA-seq continues to fall in price and rise in throughput, sample sizes will increase and training deep models to study gene expression will become even more useful.
Already several deep learning approaches have been applied to gene expression data with varying aims. For instance, many researchers have applied unsupervised deep learning models to extract meaningful representations of gene modules or sample clusters. Denoising autoencoders have been used to cluster yeast expression microarrays into known modules representing cell cycle processes [180] and to stratify yeast strains based on chemical and mutational perturbations [181]. Shallow (one hidden layer) denoising autoencoders have also been fruitful in extracting biological insight from thousands of Pseudomonas aeruginosa experiments [182,183] and in aggregating features relevant to specific breast cancer subtypes [26]. These unsupervised approaches applied to gene expression data are powerful methods for identifying gene signatures that may otherwise be overlooked. An additional benefit of unsupervised approaches is that ground truth labels, which are often difficult to acquire or are incorrect, are nonessential. However, the genes that have been aggregated into features must be interpreted carefully. Attributing each node to a single specific biological function risks over-interpreting models. Batch effects could cause models to discover non-biological features, and downstream analyses should take this into consideration.
Deep learning approaches are also being applied to gene expression prediction tasks. For example, a deep neural network with three hidden layers outperformed linear regression in inferring the expression of over 20,000 target genes based on a representative, well-connected set of about 1,000 landmark genes [184]. However, while the deep learning model outperformed existing algorithms in nearly every scenario, the model still displayed poor performance. The paper was also limited by computational bottlenecks that required data to be split randomly into two distinct models and trained separately. It is unclear how much performance would have increased if not for computational restrictions.
Epigenomic data, combined with deep learning, may have sufficient explanatory power to infer gene expression. For instance, the DeepChrome CNN [185] improved prediction accuracy of high or low gene expression from histone modifications over existing methods. AttentiveChrome [186] added a deep attention model to further enhance DeepChrome. Deep learning can also integrate different data types. For example, Liang et al. combined RBMs to integrate gene expression, DNA methylation, and miRNA data to define ovarian cancer subtypes [187]. While these approaches are promising, many convert gene expression measurements to categorical or binary variables, thus ablating many complex gene expression signatures present in intermediate and relative numbers.
Deep learning applied to gene expression data is still in its infancy, but the future is bright. Many previously untestable hypotheses can now be interrogated as deep learning enables analysis of increasing amounts of data generated by new technologies. For example, the effects of cellular heterogeneity on basic biology and disease etiology can now be explored by single-cell RNA-seq and high-throughput fluorescence-based imaging, techniques we discuss below that will benefit immensely from deep learning approaches.
DNA methylation is the process of adding a methyl group to a cytosine in the context of a CpG dinucleotide. This DNA-level epigenetic modification regulates gene transcription and is critical in development. Alterations to DNA methylation are well-established as contributing to pathophysiology of many diseases including cancers [188,189]. Studies of DNA methylation have demonstrated its fundamental role in cell lineage specification starting with stem cell differentiation [190,191] as well as a strong relationship with aging phenotypes [192,193] and pathogenesis in response to environmental exposures [194,195].
Traditional analytic approaches to DNA methylation data often focus on estimating differential DNA methylation between groups or related with an outcome using linear mixed effects models, so-called epigenome-wide association studies [196,197,198,199]. In addition, a growing application of DNA methylation measures is to infer cellular or subject phenotypes from samples and either examine the relation of these phenotypes with outcomes or disease states directly or include them in models as covariates [200,201,202,203,204]. For example, inference of subject age using DNA methylation clock approaches are established [205] and are starting to be applied to test the relation of biological age with disease risk and outcomes [206]. Different cell types have different DNA methylation profiles. A novel approach to immunophenotyping combines measurements with reference DNA methylation profiles of leukocytes to infer immune cell type proportions [207,208]. This strategy is particularly helpful when only DNA is available from a sample. Cell type inference is important for adjusting for cell-type composition in epigenome-wide association studies [199]. While reference-based libraries have strong predictive value for immune cell type estimation and have broad utility, methods to incorporate estimates of mixtures pose important considerations on the interpretation of underlying biology associated with disease manifestations and phenotypes. When a reference library is not available, reference-free deconvolution methods [209] that do not rely on these reference libraries are available to decompose signal purported to be contributed by cell types. However, using reference-free cell type proportion estimates as potential confounders in adjusted models can be overly conservative. Outcome-associated variation in DNA methylation may be decomposed into putative cell type estimates. Additional validated reference-based libraries for other tissue types, advancements in reference-free deconvolution methods, and application of deep learning methods are expected to provide new opportunities to understand and interpret DNA methylation in human health and disease.
Deep learning approaches have numerous potential applications for DNA methylation data. Imputation methods that capture complex interactions between different regions of DNA can expand the number of CpG sites whose DNA methylation state can be studied. Ideally these methods can derive their own informative, biologically-relevant features. The primary deep learning methods developed to date focus on: 1) estimating regions of methylation status and imputing missing methylation values, 2) performing classification and regression tasks, and 3) using the latent embeddings of methylation states to derive biologically meaningful features, infer interpolated disease states, and uncover CpG sites that aid the above prediction tasks.
Deep learning approaches are beginning to help address some of the current limitations of feature-by-feature analysis approaches to DNA methylation data and may help uncover additional important features necessary to understand the biological underpinnings behind different pathological states. One of the more popular applications is imputing the degree of methylation at CpG sites that are within a few thousand base pairs of measured sites or present in similar samples. DeepSignal employs a CNN to construct features from raw electrical Nanopore signals from sites near a methylated base. It uses a bidirectional RNN on DNA sequences of the aligned signals to detect methylation [210]. DeepCpG applies a similar method using scBS-Seq, DNA sequence, and a bidirectional gated recurrent network [211]. Methods like MRCNN and DeepMethyl incorporate both sequence and topological structure [212,213,214,215]. In addition, gene expression has been used to infer and impute methylation states [216,217], methylation of genes can be predicted from promoter methylation [218], and convolutional models have been able to predict methylation status from images [219,220]. While these examples of methylation imputation and inference methods have value, it is imperative to recognize limitations of imputing cytosine modifications. Imputing DNA methylation has complexities above and beyond genotype imputation. Correlation of DNA methylation marks can depend on cell types and other factors that vary by sample. As the number of tissue types and cell types with whole-genome bisulfite sequencing and oxidative bisulfite sequencing grows, the accuracy of DNA methylation imputation is expected to increase. While these methods, such as the autoencoder-based DAPL [221], reduce the computational overhead at comparable performance to other popular methylation imputation methods such as k-nearest neighbors, random forests, singular value decomposition, and multiple imputation by chained equations, the software implementations will need to become more user-friendly to gain widespread adoption.
Once DNA methylation is measured, deep learning approaches can also be used to perform classification and regression tasks. For instance, deep neural networks have been employed on DNA methylation data to predict triglyceride concentrations pre- and post-treatment [222,223] and differentiate cancer subtypes [224,225] better than other methods such as support vector machines (SVMs). Modular approaches to methylation prediction, such as MethylNet, have been able to predict age, cellular proportions, and cancer subtypes, outperforming SVM and elastic net models while remaining concordant with expected biology [226]. These approaches aim to make embedding, hyperparameter selection, regression, classification, and model interpretation tasks more tractable for epigenetics researchers and machine learning scientists.
Unsupervised discovery of biologically-significant features is another major area of interest for researchers using DNA methylation data. A consistent theme of these methods is that they construct a low-dimensional space that semantically encodes biologically important features from methylation profiles. As with other applications, these low-dimensional representations are thought to capture a set of important, unmeasured sources of biological variability in the data. Projection into these spaces results in biologically-similar examples being close together. For this reason, they are often termed latent spaces. One method used several stacked binary RBMs to learn a low-dimensional subspace representation of the methylation profiles of 5,000 CpG sites with the highest variance across 136 breast tissue samples, 113 breast cancer samples, and 23 non-cancerous samples. Samples in the latent space were clustered via self-organizing maps to show that the latent space could differentiate breast cancer samples from non-neoplastic samples. Furthermore, the latent space was visualized using t-Distributed Stochastic Neighbor Embedding (t-SNE) [227,228]. Titus et al. [229] adapted a VAE strategy developed by Way et al. [230] to methylation data. The VAE was modified to perform dimensionality reduction on 300,000 PAM50-assigned CpG features to 100 latent features in 862 samples. The authors performed t-SNE visualization, clustering, and tumor subtype classification from a TCGA breast cancer dataset. In an subsequent extension [231], the authors constructed a 100-dimensional latent space of 100,000 CpG sites across approximately 1,200 samples. They selected latent space dimensions that were the most highly associated with the differentiation between estrogen receptor (ER) positive and negative tumor samples in breast cancer patients to determine the extent to which the latent space could predict responses to endocrine therapy. Certain latent space dimensions differentiated tumors based on their ER status and provided biologically-plausible hypotheses, which suggests that VAE-derived models may have a place in summarizing DNA methylation profiles into composite features that can aid in predicting treatment response. Another study explored the latent features of lung cancer methylation profiles that were extracted using VAEs. After constructing a latent space representations of TCGA lung cancer samples, the authors used a logistic regression classifier on the latent dimensions to accurately classify cancer subtypes [232]. These studies, along with the growing body of work using VAEs and other latent representations of genomic and epigenomic data demonstrate a suite of tools to explore the unmeasured aspects of biology. Techniques that produce these representations provide the opportunity to discover important biological features that were previously missed. The power of unsupervised deep learning models for this task comes from their ability to learn high-dimensional non-linear relationships among data.
Important applications in the future include predicting methylation and pathological states based on methylation profiles uncovered from datasets with more noise, such as solid tissue samples. Unsupervised deep learning approaches such as VAEs may provide a more complete understanding of the biological processes underlying cell types, transitions in cell dynamics, and subject phenotypes. In addition, latent representations may assist with biological hypothesis generation and have the ability to stratify patients by predicted risk. While neural network embeddings can outperform traditional embeddings, it is important to be aware that many of these methods can be highly sensitive to hyperparameter tuning and an evaluation of the impact of hyperparameter tuning should be included [233].
Pre-mRNA transcripts can be spliced into different isoforms by retaining or skipping subsets of exons or including parts of introns, creating enormous spatiotemporal flexibility to generate multiple distinct proteins from a single gene. This remarkable complexity can lend itself to defects that underlie many diseases. For instance, splicing mutations in the lamin A (LMNA) gene can lead to specific variants of dilated cardiomyopathy and limb girdle muscular dystrophy [234]. A recent study found that quantitative trait loci that affect splicing in lymphoblastoid cell lines are enriched within risk loci for schizophrenia, multiple sclerosis, and other immune diseases, implicating mis-splicing as a more widespread feature of human pathologies than previously thought [235]. Therapeutic strategies that aim to modulate splicing are also currently being considered for disorders such as Duchenne muscular dystrophy and spinal muscular atrophy [234].
Sequencing studies routinely return thousands of unannotated variants, but which cause functional changes in splicing and how are those changes manifested? Prediction of a “splicing code” has been a goal of the field for the past decade. Initial machine learning approaches used a naïve Bayes model and a 2-layer Bayesian neural network with thousands of hand-derived sequence-based features to predict the probability of exon skipping [236,237]. With the advent of deep learning, more complex models provided better predictive accuracy [238,239]. Importantly, these new approaches can take in multiple kinds of epigenomic measurements as well as tissue identity and RNA binding partners of splicing factors. Deep learning is critical in furthering these kinds of integrative studies where different data types and inputs interact in unpredictable (often nonlinear) ways to create higher-order features. Moreover, as in gene expression network analysis, interrogating the hidden nodes within neural networks could potentially illuminate important aspects of splicing behavior. For instance, tissue-specific splicing mechanisms could be inferred by training networks on splicing data from different tissues, then searching for common versus distinctive hidden nodes, a technique employed by Qin et al. for tissue-specific transcription factor (TF) binding predictions [240].
A parallel effort has been to use more data with simpler models. An exhaustive study using readouts of splicing for millions of synthetic intronic sequences uncovered motifs that influence the strength of alternative splice sites [241]. The authors built a simple linear model using hexamer motif frequencies that successfully generalized to exon skipping. In a limited analysis using single nucleotide polymorphisms (SNPs) from three genes, it predicted exon skipping with three times the accuracy of an existing deep learning-based framework [238]. This case is instructive in that clever sources of data, not just more descriptive models, are still critical.
We already understand how mis-splicing of a single gene can cause diseases such as limb girdle muscular dystrophy. The challenge now is to uncover how genome-wide alternative splicing underlies complex, non-Mendelian diseases such as autism, schizophrenia, Type 1 diabetes, and multiple sclerosis [242]. As a proof of concept, Xiong et al. [238] sequenced five autism spectrum disorder and 12 control samples, each with an average of 42,000 rare variants, and identified mis-splicing in 19 genes with neural functions. Such methods may one day enable scientists and clinicians to rapidly profile thousands of unannotated variants for functional effects on splicing and nominate candidates for further investigation. Moreover, these nonlinear algorithms can deconvolve the effects of multiple variants on a single splice event without the need to perform combinatorial in vitro experiments. The ultimate goal is to predict an individual’s tissue-specific, exon-specific splicing patterns from their genome sequence and other measurements to enable a new branch of precision diagnostics that also stratifies patients and suggests targeted therapies to correct splicing defects. However, to achieve this we expect that methods to interpret the “black box” of deep neural networks and integrate diverse data sources will be required.
Transcription factors are proteins that bind regulatory DNA in a sequence-specific manner to modulate the activation and repression of gene transcription. High-throughput in vitro experimental assays that quantitatively measure the binding specificity of a TF to a large library of short oligonucleotides [243] provide rich datasets to model the naked DNA sequence affinity of individual TFs in isolation. However, in vivo TF binding is affected by a variety of other factors beyond sequence affinity, such as competition and cooperation with other TFs, TF concentration, and chromatin state (chemical modifications to DNA and other packaging proteins that DNA is wrapped around) [243]. TFs can thus exhibit highly variable binding landscapes across the same genomic DNA sequence across diverse cell types and states. Several experimental approaches such as chromatin immunoprecipitation followed by sequencing (ChIP-seq) have been developed to profile in vivo binding maps of TFs [243]. Large reference compendia of ChIP-seq data are now freely available for a large collection of TFs in a small number of reference cell states in humans and a few other model organisms [244]. Due to fundamental material and cost constraints, it is infeasible to perform these experiments for all TFs in every possible cellular state and species. Hence, predictive computational models of TF binding are essential to understand gene regulation in diverse cellular contexts.
Several machine learning approaches have been developed to learn generative and discriminative models of TF binding from in vitro and in vivo TF binding datasets that associate collections of synthetic DNA sequences or genomic DNA sequences to binary labels (bound/unbound) or continuous measures of binding. The most common class of TF binding models in the literature are those that only model the DNA sequence affinity of TFs from in vitro and in vivo binding data. The earliest models were based on deriving simple, compact, interpretable sequence motif representations such as position weight matrices (PWMs) and other biophysically inspired models [245,246,247]. These models were outperformed by general k-mer based models including SVMs with string kernels [248,249].
In 2015, Alipanahi et al. developed DeepBind, the first CNN to classify bound DNA sequences based on in vitro and in vivo assays against random DNA sequences matched for dinucleotide sequence composition [250]. The convolutional layers learn pattern detectors reminiscent of PWMs from a one-hot encoding of the raw input DNA sequences. DeepBind outperformed several state-of-the-art methods from the DREAM5 in vitro TF-DNA motif recognition challenge [247]. Although DeepBind was also applied to RNA-binding proteins, in general RNA binding is a separate problem [251] and accurate models will need to account for RNA secondary structure. Following DeepBind, several optimized convolutional and recurrent neural network architectures as well as novel hybrid approaches that combine kernel methods with neural networks have been proposed that further improve performance [252,253,254,255]. Specialized layers and regularizers have also been proposed to reduce parameters and learn more robust models by taking advantage of specific properties of DNA sequences such as their reverse complement equivalence [256,257].
While most of these methods learn independent models for different TFs, in vivo multiple TFs compete or cooperate to occupy DNA binding sites, resulting in complex combinatorial co-binding landscapes. To take advantage of this shared structure in in vivo TF binding data, multi-task neural network architectures have been developed that explicitly share parameters across models for multiple TFs [255,258,259]. Some of these multi-task models train and evaluate classification performance relative to an unbound background set of regulatory DNA sequences sampled from the genome rather than using synthetic background sequences with matched dinucleotide composition.
The above-mentioned TF binding prediction models that use only DNA sequences as inputs have a fundamental limitation. Because the DNA sequence of a genome is the same across different cell types and states, a sequence-only model of TF binding cannot predict different in vivo TF binding landscapes in new cell types not used during training. One approach for generalizing TF binding predictions to new cell types is to learn models that integrate DNA sequence inputs with other cell-type-specific data modalities that modulate in vivo TF binding such as surrogate measures of TF concentration (e.g. TF gene expression) and chromatin state. Arvey et al. showed that combining the predictions of SVMs trained on DNA sequence inputs and cell-type specific DNase-seq data, which measures genome-wide chromatin accessibility, improved in vivo TF binding prediction within and across cell types [260]. Several “footprinting” based methods have also been developed that learn to discriminate bound from unbound instances of known canonical motifs of a target TF based on high-resolution footprint patterns of chromatin accessibility that are specific to the target TF [261]. However, the genome-wide predictive performance of these methods in new cell types and states has not been evaluated.
Recently, a community challenge known as the “ENCODE-DREAM in vivo TF Binding Site Prediction Challenge” was introduced to systematically evaluate genome-wide performance of methods that can predict TF binding across cell states by integrating DNA sequence and in vitro DNA shape with cell-type-specific chromatin accessibility and gene expression [262]. A deep learning model called FactorNet was amongst the top three performing methods in the challenge [263]. FactorNet uses a multi-modal hybrid convolutional and recurrent architecture that integrates DNA sequence with chromatin accessibility profiles, gene expression, and evolutionary conservation of sequence. It is worth noting that FactorNet was slightly outperformed by an approach that does not use neural networks [264]. This top ranking approach uses an extensive set of curated features in a weighted variant of a discriminative maximum conditional likelihood model in combination with a novel iterative training strategy and model stacking. There appears to be significant room for improvement because none of the current approaches for cross cell type prediction explicitly account for the fact that TFs can co-bind with distinct co-factors in different cell states. In such cases, sequence features that are predictive of TF binding in one cell state may be detrimental to predicting binding in another.
Singh et al. developed transfer string kernels for SVMs for cross-context TF binding [265]. Domain adaptation methods that allow training neural networks which are transferable between differing training and test set distributions of sequence features could be a promising avenue going forward [266,267]. These approaches may also be useful for transferring TF binding models across species.
Another class of imputation-based cross cell type in vivo TF binding prediction methods leverage the strong correlation between combinatorial binding landscapes of multiple TFs. Given a partially complete panel of binding profiles of multiple TFs in multiple cell types, a deep learning method called TFImpute learns to predict the missing binding profile of a target TF in some target cell type in the panel based on the binding profiles of other TFs in the target cell type and the binding profile of the target TF in other cell types in the panel [240]. However, TFImpute cannot generalize predictions beyond the training panel of cell types and requires TF binding profiles of related TFs.
It is worth noting that TF binding prediction methods in the literature based on neural networks and other machine learning approaches choose to sample the set of bound and unbound sequences in a variety of different ways. These choices and the choice of performance evaluation measures significantly confound systematic comparison of model performance (see Discussion).
Several methods have also been developed to interpret neural network models of TF binding. Alipanahi et al. visualize convolutional filters to obtain insights into the sequence preferences of TFs [250]. They also introduced in silico mutation maps for identifying important predictive nucleotides in input DNA sequences by exhaustively forward propagating perturbations to individual nucleotides to record the corresponding change in output prediction. Shrikumar et al. [268] proposed efficient backpropagation based approaches to simultaneously score the contribution of all nucleotides in an input DNA sequence to an output prediction. Lanchantin et al. [253] developed tools to visualize TF motifs learned from TF binding site classification tasks. These and other general interpretation techniques (see Discussion) will be critical to improve our understanding of the biologically meaningful patterns learned by deep learning models of TF binding.
Multiple TFs act in concert to coordinate changes in gene regulation at the genomic regions known as promoters and enhancers. Each gene has an upstream promoter, essential for initiating that gene’s transcription. The gene may also interact with multiple enhancers, which can amplify transcription in particular cellular contexts. These contexts include different cell types in development or environmental stresses.
Promoters and enhancers provide a nexus where clusters of TFs and binding sites mediate downstream gene regulation, starting with transcription. The gold standard to identify an active promoter or enhancer requires demonstrating its ability to affect transcription or other downstream gene products. Even extensive biochemical TF binding data has thus far proven insufficient on its own to accurately and comprehensively locate promoters and enhancers. We lack sufficient understanding of these elements to derive a mechanistic “promoter code” or “enhancer code”. But extensive labeled data on promoters and enhancers lends itself to probabilistic classification. The complex interplay of TFs and chromatin leading to the emergent properties of promoter and enhancer activity seems particularly apt for representation by deep neural networks.
Despite decades of work, computational identification of promoters remains a stubborn problem [269]. Researchers have used neural networks for promoter recognition as early as 1996 [270]. Recently, a CNN recognized promoter sequences with sensitivity and specificity exceeding 90% [271]. Most activity in computational prediction of regulatory regions, however, has moved to enhancer identification. Because one can identify promoters with straightforward biochemical assays [272,273], the direct rewards of promoter prediction alone have decreased. But the reliable ground truth provided by these assays makes promoter identification an appealing test bed for deep learning approaches that can also identify enhancers.
Recognizing enhancers presents additional challenges. Enhancers may be up to 1,000,000 bp away from the affected promoter, and even within introns of other genes [274]. Enhancers do not necessarily operate on the nearest gene and may affect multiple genes. Their activity is frequently tissue- or context-specific. No biochemical assay can reliably identify all enhancers. Distinguishing them from other regulatory elements remains difficult, and some believe the distinction somewhat artificial [275]. While these factors make the enhancer identification problem more difficult, they also make a solution more valuable.
Several neural network approaches yielded promising results in enhancer prediction. Both Basset [276] and DeepEnhancer [277] used CNNs to predict enhancers. DECRES used a feed-forward neural network [278] to distinguish between different kinds of regulatory elements, such as active enhancers, and promoters. DECRES had difficulty distinguishing between inactive enhancers and promoters. They also investigated the power of sequence features to drive classification, finding that beyond CpG islands, few were useful.
Comparing the performance of enhancer prediction methods illustrates the problems in using metrics created with different benchmarking procedures. Both the Basset and DeepEnhancer studies include comparisons to a baseline SVM approach, gkm-SVM [249]. The Basset study reports gkm-SVM attains a mean area under the precision-recall curve (AUPR) of 0.322 over 164 cell types [276]. The DeepEnhancer study reports for gkm-SVM a dramatically different AUPR of 0.899 on nine cell types [277]. This large difference means it’s impossible to directly compare the performance of Basset and DeepEnhancer based solely on their reported metrics. DECRES used a different set of metrics altogether. To drive further progress in enhancer identification, we must develop a common and comparable benchmarking procedure (see Discussion).
In addition to the location of enhancers, identifying enhancer-promoter interactions in three-dimensional space will provide critical knowledge for understanding transcriptional regulation. SPEID used a CNN to predict these interactions with only sequence and the location of putative enhancers and promoters along a one-dimensional chromosome [279]. It compared well to other methods using a full complement of biochemical data from ChIP-seq and other epigenomic methods. Of course, the putative enhancers and promoters used were themselves derived from epigenomic methods. But one could easily replace them with the output of one of the enhancer or promoter prediction methods above.
Prediction of microRNAs (miRNAs) and miRNA targets is of great interest, as they are critical components of gene regulatory networks and are often conserved across great evolutionary distance [280,281]. While many machine learning algorithms have been applied to these tasks, they currently require extensive feature selection and optimization. For instance, one of the most widely adopted tools for miRNA target prediction, TargetScan, trained multiple linear regression models on 14 hand-curated features including structural accessibility of the target site on the mRNA, the degree of site conservation, and predicted thermodynamic stability of the miRNA-mRNA complex [282]. Some of these features, including structural accessibility, are imperfect or empirically derived. In addition, current algorithms suffer from low specificity [283].
As in other applications, deep learning promises to achieve equal or better performance in predictive tasks by automatically engineering complex features to minimize an objective function. Two recently published tools use different recurrent neural network-based architectures to perform miRNA and target prediction with solely sequence data as input [283,284]. Though the results are preliminary and still based on a validation set rather than a completely independent test set, they were able to predict microRNA target sites with higher specificity and sensitivity than TargetScan. Excitingly, these tools seem to show that RNNs can accurately align sequences and predict bulges, mismatches, and wobble base pairing without requiring the user to input secondary structure predictions or thermodynamic calculations. Further incremental advances in deep learning for miRNA and target prediction will likely be sufficient to meet the current needs of systems biologists and other researchers who use prediction tools mainly to nominate candidates that are then tested experimentally.
Proteins play fundamental roles in almost all biological processes, and understanding their structure is critical for basic biology and drug development. UniProt currently has about 94 million protein sequences, yet fewer than 100,000 proteins across all species have experimentally-solved structures in Protein Data Bank (PDB). As a result, computational structure prediction is essential for a majority of proteins. However, this is very challenging, especially when similar solved structures, called templates, are not available in PDB. Over the past several decades, many computational methods have been developed to predict aspects of protein structure such as secondary structure, torsion angles, solvent accessibility, inter-residue contact maps, disorder regions, and side-chain packing. In recent years, multiple deep learning architectures have been applied, including deep belief networks, LSTMs, CNNs, and deep convolutional neural fields (DeepCNFs) [31,285].
Here we focus on deep learning methods for two representative sub-problems: secondary structure prediction and contact map prediction. Secondary structure refers to local conformation of a sequence segment, while a contact map contains information on all residue-residue contacts. Secondary structure prediction is a basic problem and an almost essential module of any protein structure prediction package. Contact prediction is much more challenging than secondary structure prediction, but it has a much larger impact on tertiary structure prediction. In recent years, the accuracy of contact prediction has greatly improved [29,286,287,288].
One can represent protein secondary structure with three different states (alpha helix, beta strand, and loop regions) or eight finer-grained states. Accuracy of a three-state prediction is called Q3, and accuracy of an 8-state prediction is called Q8. Several groups [30,289,290] applied deep learning to protein secondary structure prediction but were unable to achieve significant improvement over the de facto standard method PSIPRED [291], which uses two shallow feedforward neural networks. In 2014, Zhou and Troyanskaya demonstrated that they could improve Q8 accuracy by using a deep supervised and convolutional generative stochastic network [292]. In 2016 Wang et al. developed a DeepCNF model that improved Q3 and Q8 accuracy as well as prediction of solvent accessibility and disorder regions [31,285]. DeepCNF achieved a higher Q3 accuracy than the standard maintained by PSIPRED for more than 10 years. This improvement may be mainly due to the ability of convolutional neural fields to capture long-range sequential information, which is important for beta strand prediction. Nevertheless, the improvements in secondary structure prediction from DeepCNF are unlikely to result in a commensurate improvement in tertiary structure prediction since secondary structure mainly reflects coarse-grained local conformation of a protein structure.
Protein contact prediction and contact-assisted folding (i.e. folding proteins using predicted contacts as restraints) represents a promising new direction for ab initio folding of proteins without good templates in PDB. Co-evolution analysis is effective for proteins with a very large number (>1000) of sequence homologs [288], but fares poorly for proteins without many sequence homologs. By combining co-evolution information with a few other protein features, shallow neural network methods such as MetaPSICOV [286] and CoinDCA-NN [293] have shown some advantage over pure co-evolution analysis for proteins with few sequence homologs, but their accuracy is still far from satisfactory. In recent years, deeper architectures have been explored for contact prediction, such as CMAPpro [294], DNCON [295] and PConsC [296]. However, blindly tested in the well-known CASP competitions, these methods did not show any advantage over MetaPSICOV [286].
Recently, Wang et al. proposed the deep learning method RaptorX-Contact [29], which significantly improves contact prediction over MetaPSICOV and pure co-evolution methods, especially for proteins without many sequence homologs. It employs a network architecture formed by one 1D residual neural network and one 2D residual neural network. Blindly tested in the latest CASP competition (i.e. CASP12 [297]), RaptorX-Contact ranked first in F₁ score on free-modeling targets as well as the whole set of targets. In CAMEO (which can be interpreted as a fully-automated CASP) [298], its predicted contacts were also able to fold proteins with a novel fold and only 65–330 sequence homologs. This technique also worked well on membrane proteins even when trained on non-membrane proteins [299]. RaptorX-Contact performed better mainly due to introduction of residual neural networks and exploitation of contact occurrence patterns by simultaneously predicting all the contacts in a single protein.
Taken together, ab initio folding is becoming much easier with the advent of direct evolutionary coupling analysis and deep learning techniques. We expect further improvements in contact prediction for proteins with fewer than 1000 homologs by studying new deep network architectures. The deep learning methods summarized above also apply to interfacial contact prediction for protein complexes but may be less effective since on average protein complexes have fewer sequence homologs. Beyond secondary structure and contact maps, we anticipate increased attention to predicting 3D protein structure directly from amino acid sequence and single residue evolutionary information [300].
Complementing computational prediction approaches, cryo-electron microscopy (cryo-EM) allows near-atomic resolution determination of protein models by comparing individual electron micrographs [301]. Detailed structures require tens of thousands of protein images [302]. Technological development has increased the throughput of image capture. New hardware, such as direct electron detectors, has made large-scale image production practical, while new software has focused on rapid, automated image processing.
Some components of cryo-EM image processing remain difficult to automate. For instance, in particle picking, micrographs are scanned to identify individual molecular images that will be used in structure refinement. In typical applications, hundreds of thousands of particles are necessary to determine a structure to near atomic resolution, making manual selection impractical [302]. Typical selection approaches are semi-supervised; a user will select several particles manually, and these selections will be used to train a classifier [303,304]. Now CNNs are being used to select particles in tools like DeepPicker [305] and DeepEM [306]. In addition to addressing shortcomings from manual selection, such as selection bias and poor discrimination of low-contrast images, these approaches also provide a means of full automation. DeepPicker can be trained by reference particles from other experiments with structurally unrelated macromolecules, allowing for fully automated application to new samples.
Downstream of particle picking, deep learning is being applied to other aspects of cryo-EM image processing. Statistical manifold learning has been implemented in the software package ROME to classify selected particles and elucidate the different conformations of the subject molecule necessary for accurate 3D structures [307]. These recent tools highlight the general applicability of deep learning approaches for image processing to increase the throughput of high-resolution cryo-EM.
Protein-protein interactions (PPIs) are highly specific and non-accidental physical contacts between proteins, which occur for purposes other than generic protein production or degradation [308]. Abundant interaction data have been generated in-part thanks to advances in high-throughput screening methods, such as yeast two-hybrid and affinity-purification with mass spectrometry. However, because many PPIs are transient or dependent on biological context, high-throughput methods can fail to capture a number of interactions. The imperfections and costs associated with many experimental PPI screening methods have motivated an interest in high-throughput computational prediction.
Many machine learning approaches to PPI have focused on text mining the literature [309,310], but these approaches can fail to capture context-specific interactions, motivating de novo PPI prediction. Early de novo prediction approaches used a variety of statistical and machine learning tools on structural and sequential data, sometimes with reference to the existing body of protein structure knowledge. In the context of PPIs—as in other domains—deep learning shows promise both for exceeding current predictive performance and for circumventing limitations from which other approaches suffer.
One of the key difficulties in applying deep learning techniques to binding prediction is the task of representing peptide and protein sequences in a meaningful way. DeepPPI [311] made PPI predictions from a set of sequence and composition protein descriptors using a two-stage deep neural network that trained two subnetworks for each protein and combined them into a single network. Sun et al. [312] applied autocovariances, a coding scheme that returns uniform-size vectors describing the covariance between physicochemical properties of the protein sequence at various positions. Wang et al. [313] used deep learning as an intermediate step in PPI prediction. They examined 70 amino acid protein sequences from each of which they extracted 1260 features. A stacked sparse autoencoder with two hidden layers was then used to reduce feature dimensions and noisiness before a novel type of classification vector machine made PPI predictions.
Beyond predicting whether or not two proteins interact, Du et al. [314] employed a deep learning approach to predict the residue contacts between two interacting proteins. Using features that describe how similar a protein’s residue is relative to similar proteins at the same position, the authors extracted uniform-length features for each residue in the protein sequence. A stacked autoencoder took two such vectors as input for the prediction of contact between two residues. The authors evaluated the performance of this method with several classifiers and showed that a deep neural network classifier paired with the stacked autoencoder significantly exceeded classical machine learning accuracy.
Because many studies used predefined higher-level features, one of the benefits of deep learning—automatic feature extraction—is not fully leveraged. More work is needed to determine the best ways to represent raw protein sequence information so that the full benefits of deep learning as an automatic feature extractor can be realized.
An important type of PPI involves the immune system’s ability to recognize the body’s own cells. The major histocompatibility complex (MHC) plays a key role in regulating this process by binding antigens and displaying them on the cell surface to be recognized by T cells. Due to its importance in immunity and immune response, peptide-MHC binding prediction is a useful problem in computational biology, and one that must account for the allelic diversity in MHC-encoding gene region.
Shallow, feed-forward neural networks are competitive methods and have made progress toward pan-allele and pan-length peptide representations. Sequence alignment techniques are useful for representing variable-length peptides as uniform-length features [315,316]. For pan-allelic prediction, NetMHCpan [317,318] used a pseudo-sequence representation of the MHC class I molecule, which included only polymorphic peptide contact residues. The sequences of the peptide and MHC were then represented using both sparse vector encoding and Blosum encoding, in which amino acids are encoded by matrix score vectors. A comparable method to the NetMHC tools is MHCflurry [319], a method which shows superior performance on peptides of lengths other than nine. MHCflurry adds placeholder amino acids to transform variable-length peptides to length 15 peptides. In training the MHCflurry feed-forward neural network [320], the authors imputed missing MHC-peptide binding affinities using a Gibbs sampling method, showing that imputation improves performance for data-sets with roughly 100 or fewer training examples. MHCflurry’s imputation method increases its performance on poorly characterized alleles, making it competitive with NetMHCpan for this task. Kuksa et al. [321] developed a shallow, higher-order neural network (HONN) comprised of both mean and covariance hidden units to capture some of the higher-order dependencies between amino acid locations. Pre-training this HONN with a semi-restricted Boltzmann machine, the authors found that the performance of the HONN exceeded that of a simple deep neural network, as well as that of NetMHC.
Deep learning’s unique flexibility was recently leveraged by Bhattacharya et al. [322], who used a gated RNN method called MHCnuggets to overcome the difficulty of multiple peptide lengths. Under this framework, they used smoothed sparse encoding to represent amino acids individually. Because MHCnuggets had to be trained for every MHC allele, performance was far better for alleles with abundant, balanced training data. Vang et al. [323] developed HLA-CNN, a method which maps amino acids onto a 15-dimensional vector space based on their context relation to other amino acids before making predictions with a CNN. In a comparison of several current methods, Bhattacharya et al. found that the top methods—NetMHC, NetMHCpan, MHCflurry, and MHCnuggets—showed comparable performance, but large differences in speed. Convolutional neural networks (in this case, HLA-CNN) showed comparatively poor performance, while shallow and recurrent neural networks performed the best. They found that MHCnuggets—the recurrent neural network—was by far the fastest-training among the top performing methods.
Because interacting proteins are more likely to share a similar function, the connectivity of a PPI network itself can be a valuable information source for the prediction of protein function [324]. To incorporate higher-order network information, it is necessary to find a lower-level embedding of network structure that preserves this higher-order structure. Rather than use hand-crafted network features, deep learning shows promise for the automatic discovery of predictive features within networks. For example, Navlakha [325] showed that a deep autoencoder was able to compress a graph to 40% of its original size, while being able to reconstruct 93% of the original graph’s edges, improving upon standard dimension reduction methods. To achieve this, each graph was represented as an adjacency matrix with rows sorted in descending node degree order, then flattened into a vector and given as input to the autoencoder. While the activity of some hidden layers correlated with several popular hand-crafted network features such as k-core size and graph density, this work showed that deep learning can effectively reduce graph dimensionality while retaining much of its structural information.
An important challenge in PPI network prediction is the task of combining different networks and types of networks. Gligorijevic et al. [326] developed a multimodal deep autoencoder, deepNF, to find a feature representation common among several different PPI networks. This common lower-level representation allows for the combination of various PPI data sources towards a single predictive task. An SVM classifier trained on the compressed features from the middle layer of the autoencoder outperformed previous methods in predicting protein function.
Hamilton et al. addressed the issue of large, heterogeneous, and changing networks with an inductive approach called GraphSAGE [327]. By finding node embeddings through learned aggregator functions that describe the node and its neighbors in the network, the GraphSAGE approach allows for the generalization of the model to new graphs. In a classification task for the prediction of protein function, Chen and Zhu [328] optimized this approach and enhanced the graph convolutional network with a preprocessing step that uses an approximation to the dropout operation. This preprocessing effectively reduces the number of graph convolutional layers and it significantly improves both training time and prediction accuracy.
A field poised for dramatic revolution by deep learning is bioimage analysis. Thus far, the primary use of deep learning for biological images has been for segmentation—that is, for the identification of biologically relevant structures in images such as nuclei, infected cells, or vasculature—in fluorescence or even brightfield channels [329]. Once so-called regions of interest have been identified, it is often straightforward to measure biological properties of interest, such as fluorescence intensities, textures, and sizes. Given the dramatic successes of deep learning in biological imaging, we simply refer to articles that review recent advancements [17,329,330]. However, user-friendly tools must be developed for deep learning to become commonplace for biological image segmentation.
We anticipate an additional paradigm shift in bioimaging that will be brought about by deep learning: what if images of biological samples, from simple cell cultures to three-dimensional organoids and tissue samples, could be mined for much more extensive biologically meaningful information than is currently standard? For example, a recent study demonstrated the ability to predict lineage fate in hematopoietic cells up to three generations in advance of differentiation [331]. In biomedical research, most often biologists decide in advance what feature to measure in images from their assay system. Although classical methods of segmentation and feature extraction can produce hundreds of metrics per cell in an image, deep learning is unconstrained by human intuition and can in theory extract more subtle features through its hidden nodes. Already, there is evidence deep learning can surpass the efficacy of classical methods [332], even using generic deep convolutional networks trained on natural images [333], known as transfer learning. Recent work by Johnson et al. [334] demonstrated how the use of a conditional adversarial autoencoder allows for a probabilistic interpretation of cell and nuclear morphology and structure localization from fluorescence images. The proposed model is able to generalize well to a wide range of subcellular localizations. The generative nature of the model allows it to produce high-quality synthetic images predicting localization of subcellular structures by directly modeling the localization of fluorescent labels. Notably, this approach reduces the modeling time by omitting the subcellular structure segmentation step.
The impact of further improvements on biomedicine could be enormous. Comparing cell population morphologies using conventional methods of segmentation and feature extraction has already proven useful for functionally annotating genes and alleles, identifying the cellular target of small molecules, and identifying disease-specific phenotypes suitable for drug screening [335,336,337]. Deep learning would bring to these new kinds of experiments—known as image-based profiling or morphological profiling—a higher degree of accuracy, stemming from the freedom from human-tuned feature extraction strategies.
Single-cell methods are generating excitement as biologists characterize the vast heterogeneity within unicellular species and between cells of the same tissue type in the same organism [338]. For instance, tumor cells and neurons can both harbor extensive somatic variation [339]. Understanding single-cell diversity in all its dimensions—genetic, epigenomic, transcriptomic, proteomic, morphologic, and metabolic—is key if treatments are to be targeted not only to a specific individual, but also to specific pathological subsets of cells. Single-cell methods also promise to uncover a wealth of new biological knowledge. A sufficiently large population of single cells will have enough representative “snapshots” to recreate timelines of dynamic biological processes. If tracking processes over time is not the limiting factor, single-cell techniques can provide maximal resolution compared to averaging across all cells in bulk tissue, enabling the study of transcriptional bursting with single-cell fluorescence in situ hybridization or the heterogeneity of epigenomic patterns with single-cell Hi-C or ATAC-seq [340,341]. Joint profiling of single-cell epigenomic and transcriptional states provides unprecedented views of regulatory processes [342].
However, large challenges exist in studying single cells. Relatively few cells can be assayed at once using current droplet, imaging, or microwell technologies, and low-abundance molecules or modifications may not be detected by chance due to a phenomenon known as dropout, not to be confused with the dropout layer of deep learning. To solve this problem, Angermueller et al. [211] trained a neural network to predict the presence or absence of methylation of a specific CpG site in single cells based on surrounding methylation signal and underlying DNA sequence, achieving several percentage points of improvement compared to random forests or deep networks trained only on CpG or sequence information. Similar deep learning methods have been applied to impute low-resolution ChIP-seq signal from bulk tissue with great success, and they could easily be adapted to single-cell data [240,343]. Deep learning has also been useful for dealing with batch effects [344].
Examining populations of single cells can reveal biologically meaningful subsets of cells as well as their underlying gene regulatory networks [345]. Unfortunately, machine learning methods generally struggle with imbalanced data—when there are many more examples of class 1 than class 2—because prediction accuracy is usually evaluated over the entire dataset. To tackle this challenge, Arvaniti et al. [346] classified healthy and cancer cells expressing 25 markers by using the most discriminative filters from a CNN trained on the data as a linear classifier. They achieved impressive performance, even for cell types where the subset percentage ranged from 0.1 to 1%, significantly outperforming logistic regression and distance-based outlier detection methods. However, they did not benchmark against random forests, which tend to work better for imbalanced data, and their data was relatively low dimensional.
Neural networks can also learn low-dimensional representations of single-cell gene expression data for visualization, clustering, and other tasks. Both scvis [347] and scVI [348] are unsupervised approaches based on variational autoencoders (VAEs). Whereas scvis primarily focuses on single-cell visualization as a replacement for t-SNE [227], the scVI model accounts for zero-inflated expression distributions and can impute zero values that are due to technical effects. Beyond VAEs, Lin et al. developed a supervised model to predict cell type [349]. Similar to transfer learning approaches for microscopy images [333], they demonstrated that the hidden layer representations were informative in general and could be used to identify cellular subpopulations or match new cells to known cell types. The supervised neural network’s representation was better overall at retrieving cell types than alternatives, but all methods struggled to recover certain cell types such as hematopoietic stem cells and inner cell mass cells. As the Human Cell Atlas [350] and related efforts generate more single-cell expression data, there will be opportunities to assess how well these low-dimensional representations generalize to new cell types as well as abundant training data to learn broadly-applicable representations.
The sheer quantity of omic information that can be obtained from each cell, as well as the number of cells in each dataset, uniquely position single-cell data to benefit from deep learning. In the future, lineage tracing could be revolutionized by using autoencoders to reduce the feature space of transcriptomic or variant data followed by algorithms to learn optimal cell differentiation trajectories [351] or by feeding cell morphology and movement into neural networks [331]. Reinforcement learning algorithms [352] could be trained on the evolutionary dynamics of cancer cells or bacterial cells undergoing selection pressure and reveal whether patterns of adaptation are random or deterministic, allowing us to develop therapeutic strategies that forestall resistance. We are excited to see the creative applications of deep learning to single-cell biology that emerge over the next few years.
Metagenomics, which refers to the study of genetic material—16S rRNA or whole-genome shotgun DNA—from microbial communities, has revolutionized the study of micro-scale ecosystems within and around us. In recent years, machine learning has proved to be a powerful tool for metagenomic analysis. 16S rRNA has long been used to deconvolve mixtures of microbial genomes, yet this ignores more than 99% of the genomic content. Subsequent tools aimed to classify 300–3000 bp reads from complex mixtures of microbial genomes based on tetranucleotide frequencies, which differ across organisms [353], using supervised [354,355] or unsupervised methods [356]. Then, researchers began to use techniques that could estimate relative abundances from an entire sample faster than classifying individual reads [357,358,359,360]. There is also great interest in identifying and annotating sequence reads [361,362]. However, the focus on taxonomic and functional annotation is just the first step. Several groups have proposed methods to determine host or environment phenotypes from the organisms that are identified [363,364,365,366] or overall sequence composition [367]. Also, researchers have looked into how feature selection can improve classification [366,368], and techniques have been proposed that are classifier-independent [369,370].
Most neural networks are used for phylogenetic classification or functional annotation from sequence data where there is ample data for training. Neural networks have been applied successfully to gene annotation (e.g. Orphelia [371] and FragGeneScan [372]). Representations (similar to word2vec [105] in natural language processing) for protein family classification have been introduced and classified with a skip-gram neural network [373]. Recurrent neural networks show good performance for homology and protein family identification [374,375].
One of the first techniques of de novo genome binning used self-organizing maps, a type of neural network [356]. Essinger et al. [376] used Adaptive Resonance Theory to cluster similar genomic fragments and showed that it had better performance than k-means. However, other methods based on interpolated Markov models [377] have performed better than these early genome binners. Neural networks can be slow and therefore have had limited use for reference-based taxonomic classification, with TAC-ELM [378] being the only neural network-based algorithm to taxonomically classify massive amounts of metagenomic data. An initial study successfully applied neural networks to taxonomic classification of 16S rRNA genes, with convolutional networks providing about 10% accuracy genus-level improvement over RNNs and random forests [379]. However, this study evaluated only 3000 sequences.
Neural network uses for classifying phenotype from microbial composition are just beginning. A simple multi-layer perceptron (MLP) was able to classify wound severity from microbial species present in the wound [380]. Recently, Ditzler et al. associated soil samples with pH level using MLPs, DBNs, and RNNs [381]. Besides classifying samples appropriately, internal phylogenetic tree nodes inferred by the networks represented features for low and high pH. Thus, hidden nodes might provide biological insight as well as new features for future metagenomic sample comparison. Also, an initial study has shown promise of these networks for diagnosing disease [382].
Challenges remain in applying deep neural networks to metagenomics problems. They are not yet ideal for phenotype classification because most studies contain tens of samples and hundreds or thousands of features (species). Such underdetermined, or ill-conditioned, problems are still a challenge for deep neural networks that require many training examples. Also, due to convergence issues [383], taxonomic classification of reads from whole genome sequencing seems out of reach at the moment for deep neural networks. There are only thousands of full-sequenced genomes as compared to hundreds of thousands of 16S rRNA sequences available for training.
However, because RNNs have been applied to base calls for the Oxford Nanopore long-read sequencer with some success [384] (discussed below), one day the entire pipeline, from denoising to functional classification, may be combined into one step using powerful LSTMs [385]. For example, metagenomic assembly usually requires binning then assembly, but could deep neural nets accomplish both tasks in one network? We believe the greatest potential in deep learning is to learn the complete characteristics of a metagenomic sample in one complex network.
While we have so far primarily discussed the role of deep learning in analyzing genomic data, deep learning can also substantially improve our ability to obtain the genomic data itself. We discuss two specific challenges: calling SNPs and indels (insertions and deletions) with high specificity and sensitivity and improving the accuracy of new types of data such as nanopore sequencing. These two tasks are critical for studying rare variation, allele-specific transcription and translation, and splice site mutations. In the clinical realm, sequencing of rare tumor clones and other genetic diseases will require accurate calling of SNPs and indels.
Current methods achieve relatively high (>99%) precision at 90% recall for SNPs and indel calls from Illumina short-read data [386], yet this leaves a large number of potentially clinically-important remaining false positives and false negatives. These methods have so far relied on experts to build probabilistic models that reliably separate signal from noise. However, this process is time consuming and fundamentally limited by how well we understand and can model the factors that contribute to noise. Recently, two groups have applied deep learning to construct data-driven unbiased noise models. One of these models, DeepVariant, leverages Inception, a neural network trained for image classification by Google Brain, by encoding reads around a candidate SNP as a 221x100 bitmap image, where each column is a nucleotide and each row is a read from the sample library [386]. The top 5 rows represent the reference, and the bottom 95 rows represent randomly sampled reads that overlap the candidate variant. Each RGBA (red/green/blue/alpha) image pixel encodes the base (A, C, G, T) as a different red value, quality score as a green value, strand as a blue value, and variation from the reference as the alpha value. The neural network outputs genotype probabilities for each candidate variant. They were able to achieve better performance than GATK [387], a leading genotype caller, even when GATK was given information about population variation for each candidate variant. Another method, still in its infancy, hand-developed 62 features for each candidate variant and fed these vectors into a fully connected deep neural network [388]. Unfortunately, this feature set required at least 15 iterations of software development to fine-tune, which suggests that these models may not generalize.
Variant calling will benefit more from optimizing neural network architectures than from developing features by hand. An interesting and informative next step would be to rigorously test if encoding raw sequence and quality data as an image, tensor, or some other mixed format produces the best variant calls. Because many of the latest neural network architectures (ResNet, Inception, Xception, and others) are already optimized for and pre-trained on generic, large-scale image datasets [389], encoding genomic data as images could prove to be a generally effective and efficient strategy.
In limited experiments, DeepVariant was robust to sequencing depth, read length, and even species [386]. However, a model built on Illumina data, for instance, may not be optimal for Pacific Biosciences long-read data or MinION nanopore data, which have vastly different specificity and sensitivity profiles and signal-to-noise characteristics. Recently, Boza et al. used bidirectional recurrent neural networks to infer the E. coli sequence from MinION nanopore electric current data with higher per-base accuracy than the proprietary hidden Markov model-based algorithm Metrichor [384]. Unfortunately, training any neural network requires a large amount of data, which is often not available for new sequencing technologies. To circumvent this, one very preliminary study simulated mutations and spiked them into somatic and germline RNA-seq data, then trained and tested a neural network on simulated paired RNA-seq and exome sequencing data [390]. Despite subsequent evaluation [391] on real somatic mutation data from the International Cancer Genome Consortium [392], further assessments are required to determine whether simulation can produce sufficiently realistic data to train reliable models.
Method development for interpreting new types of sequencing data has historically taken two steps: first, easily implemented hard cutoffs that prioritize specificity over sensitivity, then expert development of probabilistic models with hand-developed inputs [390]. We anticipate that these steps will be replaced by deep learning, which will infer features simply by its ability to optimize a complex model against data.
Artificial neural networks were originally conceived as a model for computation in the brain [7]. Although deep neural networks have evolved to become a workhorse across many fields, there is still a strong connection between deep networks and the study of the brain. The rich parallel history of artificial neural networks in computer science and neuroscience is reviewed in [393,394,395].
Convolutional neural networks were originally conceived as faithful models of visual information processing in the primate visual system, and are still considered so [396]. The activations of hidden units in consecutive layers of deep convolutional networks have been found to parallel the activity of neurons in consecutive brain regions involved in processing visual scenes. Such models of neural computation are called “encoding” models, as they predict how the nervous system might encode sensory information in the world.
Even when they are not directly modeling biological neurons, deep networks have been a useful computational tool in neuroscience. They have been developed as statistical time series models of neural activity in the brain. And in contrast to the encoding models described earlier, these models are used for decoding neural activity, for instance in brain machine interfaces [397]. They have been crucial to the field of connectomics, which is concerned with mapping the connectivity of biological neural networks in the brain. In connectomics, deep networks are used to segment the shapes of individual neurons and to infer their connectivity from 3D electron microscopic images [398], and they have also been used to infer causal connectivity from optical measurement and perturbation of neural activity [399].
It is an exciting time for neuroscience. Recent rapid progress in deep networks continues to inspire new machine learning based models of brain computation [393]. And neuroscience continues to inspire new models of artificial intelligence [395].
Given the need to make better, faster interventions at the point of care—incorporating the complex calculus of a patient’s symptoms, diagnostics, and life history—there have been many attempts to apply deep learning to patient treatment. Success in this area could help to enable personalized healthcare or precision medicine [400,401]. Earlier, we reviewed approaches for patient categorization. Here, we examine the potential for better treatment, which broadly, may be divided into methods for improved choices of interventions for patients and those for development of new interventions.
In 1996, Tu [402] compared the effectiveness of artificial neural networks and logistic regression, questioning whether these techniques would replace traditional statistical methods for predicting medical outcomes such as myocardial infarction [403] or mortality [404]. He posited that while neural networks have several advantages in representational power, the difficulties in interpretation may limit clinical applications, a limitation that still remains today. In addition, the challenges faced by physicians parallel those encountered by deep learning. For a given patient, the number of possible diseases is very large, with a long tail of rare diseases and patients are highly heterogeneous and may present with very different signs and symptoms for the same disease. Still, in 2006 Lisboa and Taktak [405] examined the use of artificial neural networks in medical journals, concluding that they improved healthcare relative to traditional screening methods in 21 of 27 studies. Recent applications of deep learning in pharmacogenomics and pharmacoepigenomics show the potential for improving patient treatment response and outcome prediction using patient-specific data, pharmacogenomic targets, and pharmacological knowledge bases [20].
While further progress has been made in using deep learning for clinical decision making, it is hindered by a challenge common to many deep learning applications: it is much easier to predict an outcome than to suggest an action to change the outcome. Several attempts [121,123] at recasting the clinical decision-making problem into a prediction problem (i.e. prediction of which treatment will most improve the patient’s health) have accurately predicted survival patterns, but technical and medical challenges remain for clinical adoption (similar to those for categorization). In particular, remaining barriers include actionable interpretability of deep learning models, fitting deep models to limited and heterogeneous data, and integrating complex predictive models into a dynamic clinical environment.
A critical challenge in providing treatment recommendations is identifying a causal relationship for each recommendation. Causal inference is often framed in terms of counterfactual question [406]. Johansson et al. [407] use deep neural networks to create representation models for covariates that capture nonlinear effects and show significant performance improvements over existing models. In a less formal approach, Kale et al. [408] first create a deep neural network to model clinical time series and then analyze the relationship of the hidden features to the output using a causal approach.
A common challenge for deep learning is the interpretability of the models and their predictions. The task of clinical decision making is necessarily risk-averse, so model interpretability is key. Without clear reasoning, it is difficult to establish trust in a model. As described above, there has been some work to directly assign treatment plans without interpretability; however, the removal of human experts from the decision-making loop make the models difficult to integrate with clinical practice. To alleviate this challenge, several studies have attempted to create more interpretable deep models, either specifically for healthcare or as a general procedure for deep learning (see Discussion).
A common application for deep learning in this domain is the temporal structure of healthcare records. Many studies [409,410,411,412] have used RNNs to categorize patients, but most stop short of suggesting clinical decisions. Nemati et al. [413] used deep reinforcement learning to optimize a heparin dosing policy for intensive care patients. However, because the ideal dosing policy is unknown, the model’s predictions must be evaluated on counter-factual data. This represents a common challenge when bridging the gap between research and clinical practice. Because the ground-truth is unknown, researchers struggle to evaluate model predictions in the absence of interventional data, but clinical application is unlikely until the model has been shown to be effective. The impressive applications of deep reinforcement learning to other domains [352] have relied on knowledge of the underlying processes (e.g. the rules of the game). Some models have been developed for targeted medical problems [414], but a generalized engine is beyond current capabilities.
A clinical deep learning task that has been more successful is the assignment of patients to clinical trials. Ithapu et al. [415] used a randomized denoising autoencoder to learn a multimodal imaging marker that predicts future cognitive and neural decline from positron emission tomography (PET), amyloid florbetapir PET, and structural magnetic resonance imaging. By accurately predicting which cases will progress to dementia, they were able to efficiently assign patients to a clinical trial and reduced the required sample sizes by a factor of five. Similarly, Artemov et al. [416] applied deep learning to predict which clinical trials were likely to fail and which were likely to succeed. By predicting the side effects and pathway activations of each drug and translating these activations to a success probability, their deep learning-based approach was able to significantly outperform a random forest classifier trained on gene expression changes. These approaches suggest promising directions to improve the efficiency of clinical trials and accelerate drug development.
Drug repositioning (or repurposing) is an attractive option for delivering new drugs to the market because of the high costs and failure rates associated with more traditional drug discovery approaches [417,418]. A decade ago, the Connectivity Map [419] had a sizeable impact. Reverse matching disease gene expression signatures with a large set of reference compound profiles allowed researchers to formulate repurposing hypotheses at scale using a simple non-parametric test. Since then, several advanced computational methods have been applied to formulate and validate drug repositioning hypotheses [420,421,422]. Using supervised learning and collaborative filtering to tackle this type of problem is proving successful, especially when coupling disease or compound omic data with topological information from protein-protein or protein-compound interaction networks [423,424,425].
For example, Menden et al. [426] used a shallow neural network to predict sensitivity of cancer cell lines to drug treatment using both cell line and drug features, opening the door to precision medicine and drug repositioning opportunities in cancer. More recently, Aliper et al. [37] used gene- and pathway-level drug perturbation transcriptional profiles from the Library of Network-Based Cellular Signatures [427] to train a fully connected deep neural network to predict drug therapeutic uses and indications. By using confusion matrices and leveraging misclassification, the authors formulated a number of interesting hypotheses, including repurposing cardiovascular drugs such as otenzepad and pinacidil for neurological disorders.
Drug repositioning can also be approached by attempting to predict novel drug-target interactions and then repurposing the drug for the associated indication [428,429]. Wang et al. [430] devised a pairwise input neural network with two hidden layers that takes two inputs, a drug and a target binding site, and predicts whether they interact. Wang et al. [38] trained individual RBMs for each target in a drug-target interaction network and used these models to predict novel interactions pointing to new indications for existing drugs. Wen et al. [39] extended this concept to deep learning by creating a DBN called DeepDTIs, which predicts interactions using chemical structure and protein sequence features.
Drug repositioning appears an obvious candidate for deep learning both because of the large amount of high-dimensional data available and the complexity of the question being asked. However, perhaps the most promising piece of work in this space [37] is more of a proof of concept than a real-world hypothesis-generation tool; notably, deep learning was used to predict drug indications but not for the actual repositioning. At present, some of the most popular state-of-the-art methods for signature-based drug repurposing [431] do not use predictive modeling. A mature and production-ready framework for drug repositioning via deep learning is currently missing.
High-throughput chemical screening in biomedical research aims to improve therapeutic options over a long term horizon [22]. The objective is to discover which small molecules (also referred to as chemical compounds or ligands) specifically affect the activity of a target, such as a kinase, protein-protein interaction, or broader cellular phenotype. This screening is often one of the first steps in a long drug discovery pipeline, where novel molecules are pursued for their ability to inhibit or enhance disease-relevant biological mechanisms [432]. Initial hits are confirmed to eliminate false positives and proceed to the lead generation stage [433], where they are evaluated for absorption, distribution, metabolism, excretion, and toxicity (ADMET) and other properties. It is desirable to advance multiple lead series, clusters of structurally-similar active chemicals, for further optimization by medicinal chemists to protect against unexpected failures in the later stages of drug discovery [432].
Computational work in this domain aims to identify sufficient candidate active compounds without exhaustively screening libraries of hundreds of thousands or millions of chemicals. Predicting chemical activity computationally is known as virtual screening. An ideal algorithm will rank a sufficient number of active compounds before the inactives, but the rankings of actives relative to other actives and inactives are less important [434]. Computational modeling also has the potential to predict ADMET traits for lead generation [435] and how drugs are metabolized [436].
Ligand-based approaches train on chemicals’ features without modeling target features (e.g. protein structure). Neural networks have a long history in this domain [21,23], and the 2012 Merck Molecular Activity Challenge on Kaggle generated substantial excitement about the potential for high-parameter deep learning approaches. The winning submission was an ensemble that included a multi-task multi-layer perceptron network [437]. The sponsors noted drastic improvements over a random forest baseline, remarking “we have seldom seen any method in the past 10 years that could consistently outperform [random forest] by such a margin” [438], but not all outside experts were convinced [439]. Subsequent work (reviewed in more detail by Goh et al. [4]) explored the effects of jointly modeling far more targets than the Merck challenge [440,441], with Ramsundar et al. [441] showing that the benefits of multi-task networks had not yet saturated even with 259 targets. Although DeepTox [442], a deep learning approach, won another competition, the Toxicology in the 21st Century (Tox21) Data Challenge, it did not dominate alternative methods as thoroughly as in other domains. DeepTox was the top performer on 9 of 15 targets and highly competitive with the top performer on the others. However, for many targets there was little separation between the top two or three methods.
The nuanced Tox21 performance may be more reflective of the practical challenges encountered in ligand-based chemical screening than the extreme enthusiasm generated by the Merck competition. A study of 22 ADMET tasks demonstrated that there are limitations to multi-task transfer learning that are in part a consequence of the degree to which tasks are related [435]. Some of the ADMET datasets showed superior performance in multi-task models with only 22 ADMET tasks compared to multi-task models with over 500 less-similar tasks. In addition, the training datasets encountered in practical applications may be tiny relative to what is available in public datasets and organized competitions. A study of BACE-1 inhibitors included only 1547 compounds [443]. Machine learning models were able to train on this limited dataset, but overfitting was a challenge and the differences between random forests and a deep neural network were negligible, especially in the classification setting. Overfitting is still a problem in larger chemical screening datasets with tens or hundreds of thousands of compounds because the number of active compounds can be very small, on the order of 0.1% of all tested chemicals for a typical target [444]. This has motivated low-parameter neural networks that emphasize compound-compound similarity, such as influence-relevance voter [434,445], instead of predicting compound activity directly from chemical features.
Much of the recent excitement in this domain has come from what could be considered a creative experimentation phase, in which deep learning has offered novel possibilities for feature representation and modeling of chemical compounds. A molecular graph, where atoms are labeled nodes and bonds are labeled edges, is a natural way to represent a chemical structure. Chemical features can be represented as a list of molecular descriptors such as molecular weight, atom counts, functional groups, charge representations, summaries of atom-atom relationships in the molecular graph, and more sophisticated derived properties [446]. Traditional machine learning approaches relied on preprocessing the graph into a feature vector of molecular descriptors or a fixed-width bit vector known as a fingerprint [447]. The same fingerprints have been used by some drug-target interaction methods discussed above [39]. An overly simplistic but approximately correct view of chemical fingerprints is that each bit represents the presence or absence of a particular chemical substructure in the molecular graph. Instead of using molecular descriptors or fingerprints as input, modern neural networks can represent chemicals as textual strings [448] or images [449] or operate directly on the molecular graph, which has enabled strategies for learning novel chemical representations.
Virtual screening and chemical property prediction have emerged as one of the major applications areas for graph-based neural networks. Duvenaud et al. [450] generalized standard circular fingerprints by substituting discrete operations in the fingerprinting algorithm with operations in a neural network, producing a real-valued feature vector instead of a bit vector. Other approaches offer trainable networks that can learn chemical feature representations that are optimized for a particular prediction task. Lusci et al. [451] applied recursive neural networks for directed acyclic graphs to undirected molecular graphs by creating an ensemble of directed graphs in which one atom is selected as the root node. Graph convolutions on undirected molecular graphs have eliminated the need to enumerate artificially directed graphs, learning feature vectors for atoms that are a function of the properties of neighboring atoms and local regions on the molecular graph [452,453,454]. More sophisticated graph algorithms [455,456] addressed limitations of standard graph convolutions that primarily operate on each node’s local neighborhood. We anticipate that these graph-based neural networks could also be applicable in other types of biological networks, such as the PPI networks we discussed previously.
Advances in chemical representation learning have also enabled new strategies for learning chemical-chemical similarity functions. Altae-Tran et al. developed a one-shot learning network [453] to address the reality that most practical chemical screening studies are unable to provide the thousands or millions of training compounds that are needed to train larger multi-task networks. Using graph convolutions to featurize chemicals, the network learns an embedding from compounds into a continuous feature space such that compounds with similar activities in a set of training tasks have similar embeddings. The approach is evaluated in an extremely challenging setting. The embedding is learned from a subset of prediction tasks (e.g. activity assays for individual proteins), and only one to ten labeled examples are provided as training data on a new task. On Tox21 targets, even when trained with one task-specific active compound and one inactive compound, the model is able to generalize reasonably well because it has learned an informative embedding function from the related tasks. Random forests, which cannot take advantage of the related training tasks, trained in the same setting are only slightly better than a random classifier. Despite the success on Tox21, performance on MUV datasets, which contains assays designed to be challenging for chemical informatics algorithms, is considerably worse. The authors also demonstrate the limitations of transfer learning as embeddings learned from the Tox21 assays have little utility for a drug adverse reaction dataset.
These novel, learned chemical feature representations may prove to be essential for accurately predicting why some compounds with similar structures yield similar target effects and others produce drastically different results. Currently, these methods are enticing but do not necessarily outperform classic approaches by a large margin. The neural fingerprints [450] were narrowly beaten by regression using traditional circular fingerprints on a drug efficacy prediction task but were superior for predicting solubility or photovoltaic efficiency. In the original study, graph convolutions [452] performed comparably to a multi-task network using standard fingerprints and slightly better than the neural fingerprints [450] on the drug efficacy task but were slightly worse than the influence-relevance voter method on an HIV dataset [434]. Broader recent benchmarking has shown that relative merits of these methods depends on the dataset and cross validation strategy [457], though evaluation in this domain often uses area under the receiver operating characteristic curve (AUROC) [458], which has limited utility due to the large class imbalance (see Discussion).
We remain optimistic for the potential of deep learning and specifically representation learning in drug discovery. Rigorous benchmarking on broad and diverse prediction tasks will be as important as novel neural network architectures to advance the state of the art and convincingly demonstrate superiority over traditional cheminformatics techniques. Fortunately, there has recently been much progress in this direction. The DeepChem software [453,459] and MoleculeNet benchmarking suite [457] built upon it contain chemical bioactivity and toxicity prediction datasets, multiple compound featurization approaches including graph convolutions, and various machine learning algorithms ranging from standard baselines like logistic regression and random forests to recent neural network architectures. Independent research groups have already contributed additional datasets and prediction algorithms to DeepChem. Adoption of common benchmarking evaluation metrics, datasets, and baseline algorithms has the potential to establish the practical utility of deep learning in chemical bioactivity prediction and lower the barrier to entry for machine learning researchers without biochemistry expertise.
One open question in ligand-based screening pertains to the benefits and limitations of transfer learning. Multi-task neural networks have shown the advantages of jointly modeling many targets [440,441]. Other studies have shown the limitations of transfer learning when the prediction tasks are insufficiently related [435,453]. This has important implications for representation learning. The typical approach to improve deep learning models by expanding the dataset size may not be applicable if only “related” tasks are beneficial, especially because task-task relatedness is ill-defined. The massive chemical state space will also influence the development of unsupervised representation learning methods [448,460]. Future work will establish whether it is better to train on massive collections of diverse compounds, drug-like small molecules, or specialized subsets.
When protein structure is available, virtual screening has traditionally relied on docking programs to predict how a compound best fits in the target’s binding site and score the predicted ligand-target complex [461]. Recently, deep learning approaches have been developed to model protein structure, which is expected to improve upon the simpler drug-target interaction algorithms described above that represent proteins with feature vectors derived from amino acid sequences [39,430].
Structure-based deep learning methods differ in whether they use experimentally-derived or predicted ligand-target complexes and how they represent the 3D structure. The Atomic CNN [462] and TopologyNet [463] models take 3D structures from PDBBind [464] as input, ensuring the ligand-target complexes are reliable. AtomNet [36] samples multiple ligand poses within the target binding site, and DeepVS [465] and Ragoza et al. [466] use a docking program to generate protein-compound complexes. If they are sufficiently accurate, these latter approaches would have wider applicability to a much larger set of compounds and proteins. However, incorrect ligand poses will be misleading during training, and the predictive performance is sensitive to the docking quality [465].
There are two established options for representing a protein-compound complex. One option, a 3D grid, can featurize the input complex [36,466]. Each entry in the grid tracks the types of protein and ligand atoms in that region of the 3D space or descriptors derived from those atoms. Alternatively, DeepVS [465] and atomic convolutions [462] offer greater flexibility in their convolutions by eschewing the 3D grid. Instead, they each implement techniques for executing convolutions over atoms’ neighboring atoms in the 3D space. Gomes et al. demonstrate that currently random forest on a 1D feature vector that describes the 3D ligand-target structure generally outperforms neural networks on the same feature vector as well as atomic convolutions and ligand-based neural networks when predicting the continuous-valued inhibition constant on the PDBBind refined dataset [462]. However, in the long term, atomic convolutions may ultimately overtake grid-based methods, as they provide greater freedom to model atom-atom interactions and the forces that govern binding affinity.
De novo drug design attempts to model the typical design-synthesize-test cycle of drug discovery in silico [467,468]. It explores an estimated 1060 synthesizable organic molecules with drug-like properties without explicit enumeration [444]. To score molecules after generation or during optimization, physics-based simulation could be used [469], but machine learning models based on techniques discussed earlier may be preferable [448], as they are much more computationally expedient. Computational efficiency is particularly important during optimization as the “scoring function” may need to be called thousands of times.
To “design” and “synthesize”, traditional de novo design software relied on classical optimizers such as genetic algorithms. These algorithms use a list of hard-coded rules to perform virtual chemical reactions on molecular structures during each iteration, leading to physically stable and synthesizable molecules [468]. Deep learning models have been proposed as an alternative. In contrast to the classical approaches, in theory generative models learned from big data would not require laboriously encoded expert knowledge to generate realistic, synthesizable molecules.
In the past few years, a large number of techniques for the generative modeling and optimization of molecules with deep learning have been explored, including RNNs, VAEs, GANs, and reinforcement learning—for a review see Elton et al. [470] or Vamathevan et al. [471].
Building off the large amount of work that has already gone into text generation [472], many generative neural networks for drug design initially represented chemicals with the simplified molecular-input line-entry system (SMILES), a standard string-based representation with characters that represent atoms, bonds, and rings [473].
The first successful demonstration of a deep learning based approach for molecular optimization occurred in 2016 with the development of a SMILES-to-SMILES autoencoder capable of learning a continuous latent feature space for molecules [448]. In this learned continuous space it is possible to interpolate between molecular structures in a manner that is not possible with discrete (e.g. bit vector or string) features or in symbolic, molecular graph space. Even more interesting is that one can perform gradient-based or Bayesian optimization of molecules within this latent space. The strategy of constructing simple, continuous features before applying supervised learning techniques is reminiscent of autoencoders trained on high-dimensional EHR data [115]. A drawback of the SMILES-to-SMILES autoencoder is that not all SMILES strings produced by the autoencoder’s decoder correspond to valid chemical structures. The Grammar Variational Autoencoder, which takes the SMILES grammar into account and is guaranteed to produce syntactically valid SMILES, helps alleviate this issue to some extent [474].
Another approach to de novo design is to train character-based RNNs on large collections of molecules, for example, ChEMBL [475], to first obtain a generic generative model for drug-like compounds [473]. These generative models successfully learn the grammar of compound representations, with 94% [476] or nearly 98% [473] of generated SMILES corresponding to valid molecular structures. The initial RNN is then fine-tuned to generate molecules that are likely to be active against a specific target by either continuing training on a small set of positive examples [473] or adopting reinforcement learning strategies [476,477]. Both the fine-tuning and reinforcement learning approaches can rediscover known, held-out active molecules.
Reinforcement learning approaches where operations are performed directly on the molecular graph bypass the need to learn the details of SMILES syntax, allowing the model to focus purely on chemistry. Additionally, they seem to require less training data and generate more valid molecules since they are constrained by design only to graph operations which satisfy chemical valiance rules [470]. A reinforcement learning agent developed by Zhou et al. [478] demonstrated superior molecular optimization performance on optimizing the quantitative estimate of drug-likeness (QED) metric and the “penalized logP” metric (logP minus the synthetic accessibility) when compared with other deep learning based approaches such as the Junction Tree VAE [479], Objective-Reinforced Generative Adversarial Network [480], and Graph Convolutional Policy Network [481]. As another example, Zhavoronkov et al. used generative tensorial reinforcement learning to discover inhibitors of discoidin domain receptor 1 (DDR1) [482]. In contrast to most previous work, six lead candidates discovered using their approach were synthesized and tested in the lab, with 4/6 achieving some degree of binding to DDR1. One of the molecules was chosen for further testing and showed promising results in a cancer cell line and mouse model [482].
In concluding this section, we want to highlight two areas where work is still needed before AI can bring added value to the existing drug discovery process—novelty and synthesizability. The work of Zhavoronkov et al. is arguably an important milestone and received much fanfare in the popular press, but Walters and Murko have presented a more sober assessment, noting that the generated molecule they choose to test in the lab is very similar to an existing drug that was present in their training data [483]. Small variations of existing molecules are likely not to be much better and may not be patentable. One way to tackle this problem is to add novelty and diversity metrics to the reward function of reinforcement learning based algorithms. Novelty should also be taken into account when comparing different models—and thus is included in the proposed GuacaMol benchmark (2019) for accessing generative molecules for molecular design [484]. The other area which has been pointed to as a key limitation of current approaches is synthesizability [485,486]. Current heuristics of synthesizability, such as the synthetic accessibility score, are based on a relatively limited domain of chemical data and are too restrictive, so better models of synthesizability should help in this area [485].
As noted before, genetic algorithms use hard-coded rules based on possible chemical reactions to generate molecular structures and therefore may have less trouble generating synthesizable molecules [468]. We note in passing that Jensen (2018) [487] and Yoshikawa et al. (2019) [488] have both demonstrated genetic algorithms that are competitive with deep learning approaches. Progress on overcoming both of these shortcomings is proceeding on many fronts, and we believe the future of deep learning for molecular design is quite bright.
Despite the disparate types of data and scientific goals in the learning tasks covered above, several challenges are broadly important for deep learning in the biomedical domain. Here we examine these factors that may impede further progress, ask what steps have already been taken to overcome them, and suggest future research directions.
Some of the challenges in applying deep learning are shared with other machine learning methods. In particular, many problem-specific optimizations described in this review reflect a recurring universal tradeoff—controlling the flexibility of a model in order to maximize predictivity. Methods for adjusting the flexibility of deep learning models include dropout, reduced data projections, and transfer learning (described below). One way of understanding such model optimizations is that they incorporate external information to limit model flexibility and thereby improve predictions. This balance is formally described as a tradeoff between “bias and variance” [11].
Although the bias-variance tradeoff is common to all machine learning applications, recent empirical and theoretical observations suggest that deep learning models may have uniquely advantageous generalization properties [489,490]. Nevertheless, additional advances will be needed to establish a coherent theoretical foundation that enables practitioners to better reason about their models from first principles.
Making predictions in the presence of high class imbalance and differences between training and generalization data is a common feature of many large biomedical datasets, including deep learning models of genomic features, patient classification, disease detection, and virtual screening. Prediction of transcription factor binding sites exemplifies the difficulties with learning from highly imbalanced data. The human genome has 3 billion base pairs, and only a small fraction of them are implicated in specific biochemical activities. Less than 1% of the genome can be confidently labeled as bound for most transcription factors.
Estimating the false discovery rate (FDR) is a standard method of evaluation in genomics that can also be applied to deep learning model predictions of genomic features. Using deep learning predictions for targeted validation experiments of specific biochemical activities necessitates a more stringent FDR (typically 5–25%). However, when predicted biochemical activities are used as features in other models, such as gene expression models, a low FDR may not be necessary.
What is the correspondence between FDR metrics and commonly used classification metrics such as AUPR and AUROC? AUPR evaluates the average precision, or equivalently, the average FDR across all recall thresholds. This metric provides an overall estimate of performance across all possible use cases, which can be misleading for targeted validation experiments. For example, classification of TF binding sites can exhibit a recall of 0% at 10% FDR and AUPR greater than 0.6. In this case, the AUPR may be competitive, but the predictions are ill-suited for targeted validation that can only examine a few of the highest-confidence predictions. Likewise, AUROC evaluates the average recall across all false positive rate (FPR) thresholds, which is often a highly misleading metric in class-imbalanced domains [72,491]. Consider a classification model with recall of 0% at FDR less than 25% and 100% recall at FDR greater than 25%. In the context of TF binding predictions where only 1% of genomic regions are bound by the TF, this is equivalent to a recall of 100% for FPR greater than 0.33%. In other words, the AUROC would be 0.9967, but the classifier would be useless for targeted validation. It is not unusual to obtain a chromosome-wide AUROC greater than 0.99 for TF binding predictions but a recall of 0% at 10% FDR. Consequently, practitioners must select the metric most tailored to their subsequent use case to use these methods most effectively.
Genome-wide continuous signals are commonly formulated into classification labels through signal peak detection. ChIP-seq peaks are used to identify locations of TF binding and histone modifications. Such procedures rely on thresholding criteria to define what constitutes a peak in the signal. This inevitably results in a set of signal peaks that are close to the threshold, not sufficient to constitute a positive label but too similar to positively labeled examples to constitute a negative label. To avoid an arbitrary label for these examples they may be labeled as “ambiguous”. Ambiguously labeled examples can then be ignored during model training and evaluation of recall and FDR. The correlation between model predictions on these examples and their signal values can be used to evaluate if the model correctly ranks these examples between positive and negative examples.
In assessing the upper bound on the predictive performance of a deep learning model, it is necessary to incorporate inherent between-study variation inherent to biomedical research [492]. Study-level variability limits classification performance and can lead to underestimating prediction error if the generalization error is estimated by splitting a single dataset. Analyses can incorporate data from multiple labs and experiments to capture between-study variation within the prediction model mitigating some of these issues.
Deep learning based solutions for biomedical applications could substantially benefit from guarantees on the reliability of predictions and a quantification of uncertainty. Due to biological variability and precision limits of equipment, biomedical data do not consist of precise measurements but of estimates with noise. Hence, it is crucial to obtain uncertainty measures that capture how noise in input values propagates through deep neural networks. Such measures can be used for reliability assessment of automated decisions in clinical and public health applications, and for guarding against model vulnerabilities in the face of rare or adversarial cases [493]. Moreover, in fundamental biological research, measures of uncertainty help researchers distinguish between true regularities in the data and patterns that are false or merely anecdotal. There are two main uncertainties that one can calculate: epistemic and aleatoric [494]. Epistemic uncertainty describes uncertainty about the model, its structure, or its parameters. This uncertainty is caused by insufficient training data or by a difference in the training set and testing set distributions, so it vanishes in the limit of infinite data. On the other hand, aleatoric uncertainty describes uncertainty inherent in the observations. This uncertainty is due to noisy or missing data, so it vanishes with the ability to observe all independent variables with infinite precision. A good way to represent aleatoric uncertainty is to design an appropriate loss function with an uncertainty variable. In the case of data-dependent aleatoric uncertainty, one can train the model to increase its uncertainty when it is incorrect due to noisy or missing data, and in the case of task-dependent aleatoric uncertainty, one can optimize for the best uncertainty parameter for each task [495]. Meanwhile, there are various methods for modeling epistemic uncertainty, outlined below.
In classification tasks, confidence calibration is the problem of using classifier scores to predict class membership probabilities that match the true membership likelihoods. These membership probabilities can be used to assess the uncertainty associated with assigning the example to each of the classes. Guo et al. [496] observed that contemporary neural networks are poorly calibrated and provided a simple recommendation for calibration: temperature scaling, a single parameter special case of Platt scaling [497]. In addition to confidence calibration, there is early work from Chryssolouris et al. [498] that described a method for obtaining confidence intervals with the assumption of normally distributed error for the neural network. More recently, Hendrycks and Gimpel discovered that incorrect or out-of-distribution examples usually have lower maximum softmax probabilities than correctly classified examples, allowing for effective detection of misclassified examples [499]. Liang et al. used temperature scaling and small perturbations to further separate the softmax scores of correctly classified examples and the scores of out-of-distribution examples, allowing for more effective detection [500]. This approach outperformed the baseline approaches by a large margin, establishing a new state-of-the-art performance.
An alternative approach for obtaining principled uncertainty estimates from deep learning models is to use Bayesian neural networks. Deep learning models are usually trained to obtain the most likely parameters given the data. However, choosing the single most likely set of parameters ignores the uncertainty about which set of parameters (among the possible models that explain the given dataset) should be used. This sometimes leads to uncertainty in predictions when the chosen likely parameters produce high-confidence but incorrect results. On the other hand, the parameters of Bayesian neural networks are modeled as full probability distributions. This Bayesian approach comes with a whole host of benefits, including better calibrated confidence estimates [501] and more robustness to adversarial and out-of-distribution examples [502]. Unfortunately, modeling the full posterior distribution for the model’s parameters given the data is usually computationally intractable. One popular method for circumventing this high computational cost is called test-time dropout [503], where an approximate posterior distribution is obtained using variational inference. Gal and Ghahramani showed that a stack of fully connected layers with dropout between the layers is equivalent to approximate inference in a Gaussian process model [503]. The authors interpret dropout as a variational inference method and apply their method to convolutional neural networks. This is simple to implement and preserves the possibility of obtaining cheap samples from the approximate posterior distribution. Operationally, obtaining model uncertainty for a given case becomes as straightforward as leaving dropout turned on and predicting multiple times. The spread of the different predictions is a reasonable proxy for model uncertainty. This technique has been successfully applied in an automated system for detecting diabetic retinopathy [504], where uncertainty-informed referrals improved diagnostic performance and allowed the model to meet the National Health Service recommended levels of sensitivity and specificity. The authors also found that entropy performs comparably to the spread obtained via test-time dropout for identifying uncertain cases, and therefore it can be used instead for automated referrals.
Several other techniques have been proposed for effectively estimating predictive uncertainty as uncertainty quantification for neural networks continues to be an active research area. Recently, McClure and Kriegeskorte observed that test-time sampling improved calibration of the probabilistic predictions, sampling weights led to more robust uncertainty estimates than sampling units, and spike-and-slab sampling was superior to Gaussian dropconnect and Bernoulli dropout [505]. Krueger et al. introduced Bayesian hypernetworks [506] as another framework for approximate Bayesian inference in deep learning, where an invertible generative hypernetwork maps isotropic Gaussian noise to parameters of the primary network allowing for computationally cheap sampling and efficient estimation of the posterior. Meanwhile, Lakshminarayanan et al. proposed using deep ensembles, which are traditionally used for boosting predictive performance, on standard (non-Bayesian) neural networks to obtain well-calibrated uncertainty estimates that are comparable to those obtained by Bayesian neural networks [507]. In cases where model uncertainty is known to be caused by a difference in training and testing distributions, domain adaptation-based techniques can help mitigate the problem [267].
Despite the success and popularity of deep learning, some deep learning models can be surprisingly brittle. Researchers are actively working on modifications to deep learning frameworks to enable them to handle probability and embrace uncertainty. Most notably, Bayesian modeling and deep learning are being integrated with renewed enthusiasm. As a result, several opportunities for innovation arise: understanding the causes of model uncertainty can lead to novel optimization and regularization techniques, assessing the utility of uncertainty estimation techniques on various model architectures and structures can be very useful to practitioners, and extending Bayesian deep learning to unsupervised settings can be a significant breakthrough [508]. Unfortunately, uncertainty quantification techniques are underutilized in the computational biology communities and largely ignored in the current deep learning for biomedicine literature. Thus, the practical value of uncertainty quantification in biomedical domains is yet to be appreciated.
As deep learning models achieve state-of-the-art performance in a variety of domains, there is a growing need to make the models more interpretable. Interpretability matters for two main reasons. First, a model that achieves breakthrough performance may have identified patterns in the data that practitioners in the field would like to understand. However, this would not be possible if the model is a black box. Second, interpretability is important for trust. If a model is making medical diagnoses, it is important to ensure the model is making decisions for reliable reasons and is not focusing on an artifact of the data. A motivating example of this can be found in Caruana et al. [509], where a model trained to predict the likelihood of death from pneumonia assigned lower risk to patients with asthma, but only because such patients were treated as higher priority by the hospital. In the context of deep learning, understanding the basis of a model’s output is particularly important as deep learning models are unusually susceptible to adversarial examples [510] and can output confidence scores over 99.99% for samples that resemble pure noise.
As the concept of interpretability is quite broad, many methods described as improving the interpretability of deep learning models take disparate and often complementary approaches.
Several approaches ascribe importance on an example-specific basis to the parts of the input that are responsible for a particular output. These can be broadly divided into perturbation-based approaches and backpropagation-based approaches.
Perturbation-based approaches change parts of the input and observe the impact on the output of the network. Alipanahi et al. [250] and Zhou & Troyanskaya [258] scored genomic sequences by introducing virtual mutations at individual positions in the sequence and quantifying the change in the output. Umarov et al. [271] used a similar strategy, but with sliding windows where the sequence within each sliding window was substituted with a random sequence. Kelley et al. [276] inserted known protein-binding motifs into the centers of sequences and assessed the change in predicted accessibility. Ribeiro et al. [511] introduced LIME, which constructs a linear model to locally approximate the output of the network on perturbed versions of the input and assigns importance scores accordingly. For analyzing images, Zeiler and Fergus [512] applied constant-value masks to different input patches. More recently, marginalizing over the plausible values of an input has been suggested as a way to more accurately estimate contributions [513].
A common drawback to perturbation-based approaches is computational efficiency: each perturbed version of an input requires a separate forward propagation through the network to compute the output. As noted by Shrikumar et al. [268], such methods may also underestimate the impact of features that have saturated their contribution to the output, as can happen when multiple redundant features are present. To reduce the computational overhead of perturbation-based approaches, Fong and Vedaldi [514] solve an optimization problem using gradient descent to discover a minimal subset of inputs to perturb in order to decrease the predicted probability of a selected class. Their method converges in many fewer iterations but requires the perturbation to have a differentiable form.
Backpropagation-based methods, in which the signal from a target output neuron is propagated backwards to the input layer, are another way to interpret deep networks that sidestep inefficiencies of the perturbation-based methods. A classic example of this is calculating the gradients of the output with respect to the input [515] to compute a “saliency map”. Bach et al. [516] proposed a strategy called Layerwise Relevance Propagation, which was shown to be equivalent to the element-wise product of the gradient and input [268,517]. Networks with Rectified Linear Units (ReLUs) create nonlinearities that must be addressed. Several variants exist for handling this [512,518]. Backpropagation-based methods are a highly active area of research. Researchers are still actively identifying weaknesses [519], and new methods are being developed to address them [520,521,268]. Lundberg and Lee [522] noted that several importance scoring methods including integrated gradients and LIME could all be considered approximations to Shapely values [523], which have a long history in game theory for assigning contributions to players in cooperative games.
Another approach to understanding the network’s predictions is to find artificial inputs that produce similar hidden representations to a chosen example. This can elucidate the features that the network uses for prediction and drop the features that the network is insensitive to. In the context of natural images, Mahendran and Vedaldi [524] introduced the “inversion” visualization, which uses gradient descent and backpropagation to reconstruct the input from its hidden representation. The method required placing a prior on the input to favor results that resemble natural images. For genomic sequence, Finnegan and Song [525] used a Markov chain Monte Carlo algorithm to find the maximum-entropy distribution of inputs that produced a similar hidden representation to the chosen input.
A related idea is “caricaturization”, where an initial image is altered to exaggerate patterns that the network searches for [526]. This is done by maximizing the response of neurons that are active in the network, subject to some regularizing constraints. Mordvintsev et al. [527] leveraged caricaturization to generate aesthetically pleasing images using neural networks.
Activation maximization can reveal patterns detected by an individual neuron in the network by generating images which maximally activate that neuron, subject to some regularizing constraints. This technique was first introduced in Ehran et al. [528] and applied in subsequent work [515,526,527,529]. Lanchantin et al. [253] applied class-based activation maximization to genomic sequence data. One drawback of this approach is that neural networks often learn highly distributed representations where several neurons cooperatively describe a pattern of interest. Thus, visualizing patterns learned by individual neurons may not always be informative.
Several interpretation methods are specifically tailored to recurrent neural network architectures. The most common form of interpretability provided by RNNs is through attention mechanisms, which have been used in diverse problems such as image captioning and machine translation to select portions of the input to focus on generating a particular output [530,531]. Deming et al. [532] applied the attention mechanism to models trained on genomic sequence. Attention mechanisms provide insight into the model’s decision-making process by revealing which portions of the input are used by different outputs. Singh et al. used a hierarchy of attention layers to locate important genome positions and signals for predicting gene expression from histone modifications [186]. In the clinical domain, Choi et al. [533] leveraged attention mechanisms to highlight which aspects of a patient’s medical history were most relevant for making diagnoses. Choi et al. [534] later extended this work to take into account the structure of disease ontologies and found that the concepts represented by the model aligned with medical knowledge. Note that interpretation strategies that rely on an attention mechanism do not provide insight into the logic used by the attention layer.
Visualizing the activation patterns of the hidden state of a recurrent neural network can also be instructive. Early work by Ghosh and Karamcheti [535] used cluster analysis to study hidden states of comparatively small networks trained to recognize strings from a finite state machine. More recently, Karpathy et al. [536] showed the existence of individual cells in LSTMs that kept track of quotes and brackets in character-level language models. To facilitate such analyses, LSTMVis [537] allows interactive exploration of the hidden state of LSTMs on different inputs.
Another strategy, adopted by Lanchatin et al. [253] looks at how the output of a recurrent neural network changes as longer and longer subsequences are supplied as input to the network, where the subsequences begin with just the first position and end with the entire sequence. In a binary classification task, this can identify those positions which are responsible for flipping the output of the network from negative to positive. If the RNN is bidirectional, the same process can be repeated on the reverse sequence. As noted by the authors, this approach was less effective at identifying motifs compared to the gradient-based backpropagation approach of Simonyan et al. [515], illustrating the need for more sophisticated strategies to assign importance scores in recurrent neural networks.
Murdoch and Szlam [538] showed that the output of an LSTM can be decomposed into a product of factors, where each factor can be interpreted as the contribution at a particular timestep. The contribution scores were then used to identify key phrases from a model trained for sentiment analysis and obtained superior results compared to scores derived via a gradient-based approach.
Interpretation of embedded or latent space features learned through generative unsupervised models can reveal underlying patterns otherwise masked in the original input. Embedded feature interpretation has been emphasized mostly in image and text based applications [105,539], but applications to genomic and biomedical domains are increasing.
For example, Way and Greene trained a VAE on gene expression from The Cancer Genome Atlas (TCGA) [540] and use latent space arithmetic to rapidly isolate and interpret gene expression features descriptive of high grade serous ovarian cancer subtypes [541]. The most differentiating VAE features were representative of biological processes that are known to distinguish the subtypes. Latent space arithmetic with features derived using other compression algorithms were not as informative in this context [542]. Embedding discrete chemical structures with autoencoders and interpreting the learned continuous representations with latent space arithmetic has also facilitated predicting drug-like compounds [448]. Furthermore, embedding biomedical text into lower dimensional latent spaces have improved name entity recognition in a variety of tasks including annotating clinical abbreviations, genes, cell lines, and drug names [78,79,80,81].
Other approaches have used interpolation through latent space embeddings learned by GANs to interpret unobserved intermediate states. For example, Osokin et al. trained GANs on two-channel fluorescent microscopy images to interpret intermediate states of protein localization in yeast cells [543]. Goldsborough et al. trained a GAN on fluorescent microscopy images and used latent space interpolation and arithmetic to reveal underlying responses to small molecule perturbations in cell lines [544].
It can often be informative to understand how the training data affects model learning. Toward this end, Koh and Liang [545] used influence functions, a technique from robust statistics, to trace a model’s predictions back through the learning algorithm to identify the datapoints in the training set that had the most impact on a given prediction. A more free-form approach to interpretability is to visualize the activation patterns of the network on individual inputs and on subsets of the data. ActiVis and CNNvis [546,547] are two frameworks that enable interactive visualization and exploration of large-scale deep learning models. An orthogonal strategy is to use a knowledge distillation approach to replace a deep learning model with a more interpretable model that achieves comparable performance. Towards this end, Che et al. [548] used gradient boosted trees to learn interpretable healthcare features from trained deep models.
Finally, it is sometimes possible to train the model to provide justifications for its predictions. Lei et al. [549] used a generator to identify “rationales”, which are short and coherent pieces of the input text that produce similar results to the whole input when passed through an encoder. The authors applied their approach to a sentiment analysis task and obtained substantially superior results compared to an attention-based method.
While deep learning lags behind most Bayesian models in terms of interpretability, the interpretability of deep learning is comparable to or exceeds that of many other widely-used machine learning methods such as random forests or SVMs. While it is possible to obtain importance scores for different inputs in a random forest, the same is true for deep learning. Similarly, SVMs trained with a nonlinear kernel are not easily interpretable because the use of the kernel means that one does not obtain an explicit weight matrix. Finally, it is worth noting that some simple machine learning methods are less interpretable in practice than one might expect. A linear model trained on heavily engineered features might be difficult to interpret as the input features themselves are difficult to interpret. Similarly, a decision tree with many nodes and branches may also be difficult for a human to make sense of.
There are several directions that might benefit the development of interpretability techniques. The first is the introduction of gold standard benchmarks that different interpretability approaches could be compared against, similar in spirit to how the ImageNet [46] and CIFAR [550] datasets spurred the development of deep learning for computer vision. It would also be helpful if the community placed more emphasis on domains outside of computer vision. Computer vision is often used as the example application of interpretability methods, but it is not the domain with the most pressing need. Finally, closer integration of interpretability approaches with popular deep learning frameworks would make it easier for practitioners to apply and experiment with different approaches to understanding their deep learning models.
A lack of large-scale, high-quality, correctly labeled training data has impacted deep learning in nearly all applications we have discussed. The challenges of training complex, high-parameter neural networks from few examples are obvious, but uncertainty in the labels of those examples can be just as problematic. In genomics labeled data may be derived from an experimental assay with known and unknown technical artifacts, biases, and error profiles. It is possible to weight training examples or construct Bayesian models to account for uncertainty or non-independence in the data, as described in the TF binding example above. As another example, Park et al. [551] estimated shared non-biological signal between datasets to correct for non-independence related to assay platform or other factors in a Bayesian integration of many datasets. However, such techniques are rarely placed front and center in any description of methods and may be easily overlooked.
For some types of data, especially images, it is straightforward to augment training datasets by splitting a single labeled example into multiple examples. For example, an image can easily be rotated, flipped, or translated and retain its label [43]. 3D MRI and 4D fMRI (with time as a dimension) data can be decomposed into sets of 2D images [552]. This can greatly expand the number of training examples but artificially treats such derived images as independent instances and sacrifices the structure inherent in the data. CellCnn trains a model to recognize rare cell populations in single-cell data by creating training instances that consist of subsets of cells that are randomly sampled with replacement from the full dataset [346].
Simulated or semi-synthetic training data has been employed in multiple biomedical domains, though many of these ideas are not specific to deep learning. Training and evaluating on simulated data, for instance, generating synthetic TF binding sites with position weight matrices [256] or RNA-seq reads for predicting mRNA transcript boundaries [553], is a standard practice in bioinformatics. This strategy can help benchmark algorithms when the available gold standard dataset is imperfect, but it should be paired with an evaluation on real data, as in the prior examples [256,553]. In rare cases, models trained on simulated data have been successfully applied directly to real data [553].
Data can be simulated to create negative examples when only positive training instances are available. DANN [35] adopts this approach to predict the pathogenicity of genetic variants using semi-synthetic training data from Combined Annotation-Dependent Depletion (CADD) [554]. Though our emphasis here is on the training strategy, it should be noted that logistic regression outperformed DANN when distinguishing known pathogenic mutations from likely benign variants in real data. Similarly, a somatic mutation caller has been trained by injecting mutations into real sequencing datasets [390,391].
In settings where the experimental observations are biased toward positive instances, such as MHC protein and peptide ligand binding affinity [320], or the negative instances vastly outnumber the positives, such as high-throughput chemical screening [445], training datasets have been augmented by adding additional instances and assuming they are negative. There is some evidence that this can improve performance [445], but in other cases it was only beneficial when the real training datasets were extremely small [320]. Overall, training with simulated and semi-simulated data is a valuable idea for overcoming limited sample sizes but one that requires more rigorous evaluation on real ground-truth datasets before we can recommend it for widespread use. There is a risk that a model will easily discriminate synthetic examples but not generalize to real data.
Multimodal, multi-task, and transfer learning, discussed in detail below, can also combat data limitations to some degree. There are also emerging network architectures, such as Diet Networks for high-dimensional SNP data [555]. These use multiple networks to drastically reduce the number of free parameters by first flipping the problem and training a network to predict parameters (weights) for each input (SNP) to learn a feature embedding. This embedding (e.g. from principal component analysis, per class histograms, or a word2vec [105] generalization) can be learned directly from input data or take advantage of other datasets or domain knowledge. Additionally, in this task the features are the examples, an important advantage when it is typical to have 500 thousand or more SNPs and only a few thousand patients. Finally, this embedding is of a much lower dimension, allowing for a large reduction in the number of free parameters. In the example given, the number of free parameters was reduced from 30 million to 50 thousand, a factor of 600.
Efficiently scaling deep learning is challenging, and there is a high computational cost (e.g. time, memory, and energy) associated with training neural networks and using them to make predictions. This is one of the reasons why neural networks have only recently found widespread use [556].
Many have sought to curb these costs, with methods ranging from the very applied (e.g. reduced numerical precision [557,558,559,560]) to the exotic and theoretic (e.g. training small networks to mimic large networks and ensembles [561,562]). The largest gains in efficiency have come from computation with GPUs [556,563,564,565,566,567], which excel at the matrix and vector operations so central to deep learning. The massively parallel nature of GPUs allows additional optimizations, such as accelerated mini-batch gradient descent [564,565,568,569]. However, GPUs also have limited memory, making networks of useful size and complexity difficult to implement on a single GPU or machine [68,563]. This restriction has sometimes forced computational biologists to use workarounds or limit the size of an analysis. Chen et al. [184] inferred the expression level of all genes with a single neural network, but due to memory restrictions they randomly partitioned genes into two separately analyzed halves. In other cases, researchers limited the size of their neural network [29] or the total number of training instances [448]. Some have also chosen to use standard central processing unit (CPU) implementations rather than sacrifice network size or performance [570].
While steady improvements in GPU hardware may alleviate this issue, it is unclear whether advances will occur quickly enough to keep pace with the growing biological datasets and increasingly complex neural networks. Much has been done to minimize the memory requirements of neural networks [557,558,559,560,561,571,572], but there is also growing interest in specialized hardware, such as field-programmable gate arrays (FPGAs) [567,573] and application-specific integrated circuits (ASICs) [574]. Less software is available for such highly specialized hardware [573]. But specialized hardware promises improvements in deep learning at reduced time, energy, and memory [567]. Specialized hardware may be a difficult investment for those not solely interested in deep learning, but for those with a deep learning focus these solutions may become popular.
Distributed computing is a general solution to intense computational requirements and has enabled many large-scale deep learning efforts. Some types of distributed computation [575,576] are not suitable for deep learning [577], but much progress has been made. There now exist a number of algorithms [559,577], tools [578,579,580], and high-level libraries [581,582] for deep learning in a distributed environment, and it is possible to train very complex networks with limited infrastructure [583]. Besides handling very large networks, distributed or parallelized approaches offer other advantages, such as improved ensembling [584] or accelerated hyperparameter optimization [585,586].
Cloud computing, which has already seen wide adoption in genomics [587], could facilitate easier sharing of the large datasets common to biology [588,589], and may be key to scaling deep learning. Cloud computing affords researchers flexibility, and enables the use of specialized hardware (e.g. FPGAs, ASICs, GPUs) without major investment. As such, it could be easier to address the different challenges associated with the multitudinous layers and architectures available [590]. Though many are reluctant to store sensitive data (e.g. patient electronic health records) in the cloud, secure, regulation-compliant cloud services do exist [591].
A robust culture of data, code, and model sharing would speed advances in this domain. The cultural barriers to data sharing in particular are perhaps best captured by the use of the term “research parasite” to describe scientists who use data from other researchers [592]. A field that honors only discoveries and not the hard work of generating useful data will have difficulty encouraging scientists to share their hard-won data. It’s precisely those data that would help to power deep learning in the domain. Efforts are underway to recognize those who promote an ecosystem of rigorous sharing and analysis [593].
The sharing of high-quality, labeled datasets will be especially valuable. In addition, researchers who invest time to preprocess datasets to be suitable for deep learning can make the preprocessing code (e.g. Basset [276] and variationanalysis [388]) and cleaned data (e.g. MoleculeNet [457]) publicly available to catalyze further research. However, there are complex privacy and legal issues involved in sharing patient data that cannot be ignored. Solving these issues will require increased understanding of privacy risks and standards specifying acceptable levels. In some domains high-quality training data has been generated privately, i.e. high-throughput chemical screening data at pharmaceutical companies. One perspective is that there is little expectation or incentive for this private data to be shared. However, data are not inherently valuable. Instead, the insights that we glean from them are where the value lies. Private companies may establish a competitive advantage by releasing data sufficient for improved methods to be developed. Recently, Ramsundar et al. did this with an open source platform DeepChem, where they released four privately generated datasets [594].
Code sharing and open source licensing is essential for continued progress in this domain. We strongly advocate following established best practices for sharing source code, archiving code in repositories that generate digital object identifiers, and open licensing [595] regardless of the minimal requirements, or lack thereof, set by journals, conferences, or preprint servers. In addition, it is important for authors to share not only code for their core models but also scripts and code used for data cleaning (see above) and hyperparameter optimization. These improve reproducibility and serve as documentation of the detailed decisions that impact model performance but may not be exhaustively captured in a manuscript’s methods text.
Because many deep learning models are often built using one of several popular software frameworks, it is also possible to directly share trained predictive models. The availability of pre-trained models can accelerate research, with image classifiers as an apt example. A pre-trained neural network can be quickly fine-tuned on new data and used in transfer learning, as discussed below. Taking this idea to the extreme, genomic data has been artificially encoded as images in order to benefit from pre-trained image classifiers [386]. “Model zoos”—collections of pre-trained models—are not yet common in biomedical domains but have started to appear in genomics applications [211,596]. However, it is important to note that sharing models trained on individual data requires great care because deep learning models can be attacked to identify examples used in training. One possible solution to protect individual samples includes training models under differential privacy [155], which has been used in the biomedical domain [158]. We discussed this issue as well as recent techniques to mitigate these concerns in the patient categorization section.
DeepChem [453,457,459] and DragoNN (Deep RegulAtory GenOmic Neural Networks) [596] exemplify the benefits of sharing pre-trained models and code under an open source license. DeepChem, which targets drug discovery and quantum chemistry, has actively encouraged and received community contributions of learning algorithms and benchmarking datasets. As a consequence, it now supports a large suite of machine learning approaches, both deep learning and competing strategies, that can be run on diverse test cases. This realistic, continual evaluation will play a critical role in assessing which techniques are most promising for chemical screening and drug discovery. Like formal, organized challenges such as the ENCODE-DREAM in vivo Transcription Factor Binding Site Prediction Challenge [262], DeepChem provides a forum for the fair, critical evaluations that are not always conducted in individual methodological papers, which can be biased toward favoring a new proposed algorithm. Likewise DragoNN offers not only code and a model zoo but also a detailed tutorial and partner package for simulating training data. These resources, especially the ability to simulate datasets that are sufficiently complex to demonstrate the challenges of training neural networks but small enough to train quickly on a CPU, are important for training students and attracting machine learning researchers to problems in genomics and healthcare.
The fact that biomedical datasets often contain a limited number of instances or labels can cause poor performance of deep learning algorithms. These models are particularly prone to overfitting due to their high representational power. However, transfer learning techniques, also known as domain adaptation, enable transfer of extracted patterns between different datasets and even domains. This approach consists of training a model for the base task and subsequently reusing the trained model for the target problem. The first step allows a model to take advantage of a larger amount of data and/or labels to extract better feature representations. Transferring learned features in deep neural networks improves performance compared to randomly initialized features even when pre-training and target sets are dissimilar. However, transferability of features decreases as the distance between the base task and target task increases [597].
In image analysis, previous examples of deep transfer learning applications proved large-scale natural image sets [46] to be useful for pre-training models that serve as generic feature extractors for various types of biological images [15,333,598,599]. More recently, deep learning models predicted protein sub-cellular localization for proteins not originally present in a training set [600]. Moreover, learned features performed reasonably well even when applied to images obtained using different fluorescent labels, imaging techniques, and different cell types [601]. However, there are no established theoretical guarantees for feature transferability between distant domains such as natural images and various modalities of biological imaging. Because learned patterns are represented in deep neural networks in a layer-wise hierarchical fashion, this issue is usually addressed by fixing an empirically chosen number of layers that preserve generic characteristics of both training and target datasets. The model is then fine-tuned by re-training top layers on the specific dataset in order to re-learn domain-specific high level concepts (e.g. fine-tuning for radiology image classification [58]). Fine-tuning on specific biological datasets enables more focused predictions.
In genomics, the Basset package [276] for predicting chromatin accessibility was shown to rapidly learn and accurately predict on new data by leveraging a model pre-trained on available public data. To simulate this scenario, authors put aside 15 of 164 cell type datasets and trained the Basset model on the remaining 149 datasets. Then, they fine-tuned the model with one training pass of each of the remaining datasets and achieved results close to the model trained on all 164 datasets together. In another example, Min et al. [277] demonstrated how training on the experimentally-validated FANTOM5 permissive enhancer dataset followed by fine-tuning on ENCODE enhancer datasets improved cell type-specific predictions, outperforming state-of-the-art results. In drug design, general RNN models trained to generate molecules from the ChEMBL database have been fine-tuned to produce drug-like compounds for specific targets [473,476].
Related to transfer learning, multimodal learning assumes simultaneous learning from various types of inputs, such as images and text. It can capture features that describe common concepts across input modalities. Generative graphical models like RBMs, deep Boltzmann machines, and DBNs, demonstrate successful extraction of more informative features for one modality (images or video) when jointly learned with other modalities (audio or text) [602]. Deep graphical models such as DBNs are well-suited for multimodal learning tasks because they learn a joint probability distribution from inputs. They can be pre-trained in an unsupervised fashion on large unlabeled data and then fine-tuned on a smaller number of labeled examples. When labels are available, convolutional neural networks are ubiquitously used because they can be trained end-to-end with backpropagation and demonstrate state-of-the-art performance in many discriminative tasks [15].
Jha et al. [239] showed that integrated training delivered better performance than individual networks. They compared a number of feed-forward architectures trained on RNA-seq data with and without an additional set of CLIP-seq, knockdown, and over-expression based input features. The integrative deep model generalized well for combined data, offering a large performance improvement for alternative splicing event estimation. Chaudhary et al. [603] trained a deep autoencoder model jointly on RNA-seq, miRNA-seq, and methylation data from TCGA to predict survival subgroups of hepatocellular carcinoma patients. This multimodal approach that treated different omic data types as different modalities outperformed both traditional methods (principal component analysis) and single-omic models. Interestingly, multi-omic model performance did not improve when combined with clinical information, suggesting that the model was able to capture redundant contributions of clinical features through their correlated genomic features. Chen et al. [179] used deep belief networks to learn phosphorylation states of a common set of signaling proteins in primary cultured bronchial cells collected from rats and humans treated with distinct stimuli. By interpreting species as different modalities representing similar high-level concepts, they showed that DBNs were able to capture cross-species representation of signaling mechanisms in response to a common stimuli. Another application used DBNs for joint unsupervised feature learning from cancer datasets containing gene expression, DNA methylation, and miRNA expression data [187]. This approach allowed for the capture of intrinsic relationships in different modalities and for better clustering performance over conventional k-means.
Multimodal learning with CNNs is usually implemented as a collection of individual networks in which each learns representations from single data type. These individual representations are further concatenated before or within fully-connected layers. FIDDLE [604] is an example of a multimodal CNN that represents an ensemble of individual networks that take NET-seq, MNase-seq, ChIP-seq, RNA-seq, and raw DNA sequence as input to predict transcription start sites. The combined model radically improves performance over separately trained datatype-specific networks, suggesting that it learns the synergistic relationship between datasets.
Multi-task learning is an approach related to transfer learning. In a multi-task learning framework, a model learns a number of tasks simultaneously such that features are shared across them. DeepSEA [258] implemented multi-task joint learning of diverse chromatin factors from raw DNA sequence. This allowed a sequence feature that was effective in recognizing binding of a specific TF to be simultaneously used by another predictor for a physically interacting TF. Similarly, TFImpute [240] learned information shared across transcription factors and cell lines to predict cell-specific TF binding for TF-cell line combinations. Yoon et al. [104] demonstrated that predicting the primary cancer site from cancer pathology reports together with its laterality substantially improved the performance for the latter task, indicating that multi-task learning can effectively leverage the commonality between two tasks using a shared representation. Many studies employed multi-task learning to predict chemical bioactivity [437,441] and drug toxicity [442,605]. Kearnes et al. [435] systematically compared single-task and multi-task models for ADMET properties and found that multi-task learning generally improved performance. Smaller datasets tended to benefit more than larger datasets.
Multi-task learning is complementary to multimodal and transfer learning. All three techniques can be used together in the same model. For example, Zhang et al. [598] combined deep model-based transfer and multi-task learning for cross-domain image annotation. One could imagine extending that approach to multimodal inputs as well. A common characteristic of these methods is better generalization of extracted features at various hierarchical levels of abstraction, which is attained by leveraging relationships between various inputs and task objectives.
Despite demonstrated improvements, transfer learning approaches pose challenges. There are no theoretically sound principles for pre-training and fine-tuning. Best practice recommendations are heuristic and must account for additional hyper-parameters that depend on specific deep architectures, sizes of the pre-training and target datasets, and similarity of domains. However, similarity of datasets and domains in transfer learning and relatedness of tasks in multi-task learning are difficult to access. Most studies address these limitations by empirical evaluation of the model. Unfortunately, negative results are typically not reported. A deep CNN trained on natural images boosts performance in radiographic images [58]. However, due to differences in imaging domains, the target task required either re-training the initial model from scratch with special pre-processing or fine-tuning of the whole network on radiographs with heavy data augmentation to avoid overfitting. Exclusively fine-tuning top layers led to much lower validation accuracy (81.4 versus 99.5). Fine-tuning the aforementioned Basset model with more than one pass resulted in overfitting [276]. DeepChem successfully improved results for low-data drug discovery with one-shot learning for related tasks. However, it clearly demonstrated the limitations of cross-task generalization across unrelated tasks in one-shot models, specifically nuclear receptor assays and patient adverse reactions [453].
In the medical domain, multimodal, multi-task and transfer learning strategies not only inherit most methodological issues from natural image, text, and audio domains, but also pose domain-specific challenges. There is a compelling need for the development of privacy-preserving transfer learning algorithms, such as Private Aggregation of Teacher Ensembles [161]. We suggest that these types of models deserve deeper investigation to establish sound theoretical guarantees and determine limits for the transferability of features between various closely related and distant learning tasks.
Deep learning-based methods now match or surpass the previous state of the art in a diverse array of tasks in patient and disease categorization, fundamental biological study, genomics, and treatment development. Returning to our central question: given this rapid progress, has deep learning transformed the study of human disease? Though the answer is highly dependent on the specific domain and problem being addressed, we conclude that deep learning has not yet realized its transformative potential or induced a strategic inflection point. Despite its dominance over competing machine learning approaches in many of the areas reviewed here and quantitative improvements in predictive performance, deep learning has not yet definitively “solved” these problems.
As an analogy, consider recent progress in conversational speech recognition. Since 2009 there have been drastic performance improvements with error rates dropping from more than 20% to less than 6% [606] and finally approaching or exceeding human performance in the past year [607,608]. The phenomenal improvements on benchmark datasets are undeniable, but greatly reducing the error rate on these benchmarks did not fundamentally transform the domain. Widespread adoption of conversational speech technologies will require solving the problem, i.e. methods that surpass human performance, and persuading users to adopt them [606]. We see parallels in healthcare, where achieving the full potential of deep learning will require outstanding predictive performance as well as acceptance and adoption by biologists and clinicians. These experts will rightfully demand rigorous evidence that deep learning has impacted their respective disciplines—elucidated new biological mechanisms and improved patient outcomes—to be convinced that the promises of deep learning are more substantive than those of previous generations of artificial intelligence.
Some of the areas we have discussed are closer to surpassing this lofty bar than others, generally those that are more similar to the non-biomedical tasks that are now monopolized by deep learning. In medical imaging, diabetic retinopathy [50], diabetic macular edema [50], tuberculosis [59], and skin lesion [5] classifiers are highly accurate and comparable to clinician performance.
In other domains, perfect accuracy will not be required because deep learning will primarily prioritize experiments and assist discovery. For example, in chemical screening for drug discovery, a deep learning system that successfully identifies dozens or hundreds of target-specific, active small molecules from a massive search space would have immense practical value even if its overall precision is modest. In medical imaging, deep learning can point an expert to the most challenging cases that require manual review [59], though the risk of false negatives must be addressed. In protein structure prediction, errors in individual residue-residue contacts can be tolerated when using the contacts jointly for 3D structure modeling. Improved contact map predictions [29] have led to notable improvements in fold and 3D structure prediction for some of the most challenging proteins, such as membrane proteins [299].
Conversely, the most challenging tasks may be those in which predictions are used directly for downstream modeling or decision-making, especially in the clinic. As an example, errors in sequence variant calling will be amplified if they are used directly for GWAS. In addition, the stochasticity and complexity of biological systems implies that for some problems, for instance predicting gene regulation in disease, perfect accuracy will be unattainable.
We are witnessing deep learning models achieving human-level performance across a number of biomedical domains. However, machine learning algorithms, including deep neural networks, are also prone to mistakes that humans are much less likely to make, such as misclassification of adversarial examples [609,610], a reminder that these algorithms do not understand the semantics of the objects presented. It may be impossible to guarantee that a model is not susceptible to adversarial examples, but work in this area is continuing [611,612]. Cooperation between human experts and deep learning algorithms addresses many of these challenges and can achieve better performance than either individually [65]. For sample and patient classification tasks, we expect deep learning methods to augment clinicians and biomedical researchers.
We are optimistic about the future of deep learning in biology and medicine. It is by no means inevitable that deep learning will revolutionize these domains, but given how rapidly the field is evolving, we are confident that its full potential in biomedicine has not been explored. We have highlighted numerous challenges beyond improving training and predictive accuracy, such as preserving patient privacy and interpreting models. Ongoing research has begun to address these problems and shown that they are not insurmountable. Deep learning offers the flexibility to model data in its most natural form, for example, longer DNA sequences instead of k-mers for transcription factor binding prediction and molecular graphs instead of pre-computed bit vectors for drug discovery. These flexible input feature representations have spurred creative modeling approaches that would be infeasible with other machine learning techniques. Unsupervised methods are currently less-developed than their supervised counterparts, but they may have the most potential because of how expensive and time-consuming it is to label large amounts of biomedical data. If future deep learning algorithms can summarize very large collections of input data into interpretable models that spur scientists to ask questions that they did not know how to ask, it will be clear that deep learning has transformed biology and medicine.
We recognized that deep learning in precision medicine is a rapidly developing area. Hence, diverse expertise was required to provide a forward-looking perspective. Accordingly, we collaboratively wrote this review in the open, enabling anyone with expertise to contribute. We wrote the manuscript in markdown and tracked changes using git. Contributions were handled through GitHub, with individuals submitting “pull requests” to suggest additions to the manuscript. This collaborative writing approach was later generalized into Manubot [613].
Manubot supports citations of persistent identifiers, such as DOIs, PubMed Central IDs, PubMed IDs, arXiv IDs, and URLs. This reduces one major barrier to writing collaboratively, which is syncing reference managers between participants. In addition, Manubot uses continuous integration to build and deploy manuscripts. This allows for automated error checking of proposed changes to catch malformated citations and invalid syntax. Originally, the Deep Review used Travis CI for continuous integration, but in 2020 switched to GitHub Actions, which became the default for Manubot manuscripts.
For version 1.0 of the Deep Review, author order was randomized as described in version 1.0 [614]. However, this was a one-time manual process. Starting with version 2.0, we began shuffling authors for every manuscript version. Manubot allowed us to automate this process, using the Git commit hash as a random seed to ensure reproducible ordering.
We continued using the open repository on the GitHub version control platform (greenelab/deep-review) [615], which was established to write the version 1.0 manuscript.
Drafted one or more subsections: Brock C. Christensen, Alexander J. Titus, Joshua J. Levy.
Drafted sub-sections, edited the manuscript, reviewed pull requests, and coordinated co-authors: Anthony Gitter, Casey S. Greene.
Edited the manuscript, reviewed pull requests, and developed Manubot: Daniel S. Himmelstein.
Revised specific sub-sections or supervised drafting one or more sub-sections: Daniel C. Elton.
| Author | Competing Interests | Last Reviewed |
|---|---|---|
| Brock C. Christensen | None | 2020-03-05 |
| Anthony Gitter | Filed a provisional patent application with the Wisconsin Alumni Research Foundation related to classifying activated T cells | 2020-08-09 |
| Daniel S. Himmelstein | None | 2020-03-10 |
| Alexander J. Titus | None | 2020-03-07 |
| Joshua J. Levy | None | 2020-03-04 |
| Casey S. Greene | None | 2020-03-10 |
| Daniel C. Elton | None | 2020-03-05 |
We acknowledge funding from the Gordon and Betty Moore Foundation award GBMF4552 (C.S.G. and D.S.H.); the National Institutes of Health awards R01HG010067 (C.S.G. and D.S.H.), R01CA237170 (C.S.G), T32LM012204 (A.J.T.), R01CA216265 (B.C.C.); the Burroughs Wellcome Fund Big Data in the Life Sciences training grant at Dartmouth (J.L.L,); and the Dartmouth College Neukom Institute for Computational Science CompX award (B.C.C.).
We created an open repository on the GitHub version control platform (greenelab/deep-review) [615].
Here, we engaged with numerous authors from papers within and outside of the area.
The manuscript was drafted via GitHub commits by individuals who met the ICMJE standards of authorship.
These were individuals who contributed to the review of the literature;
drafted the manuscript or provided substantial critical revisions;
approved the final manuscript draft; and agreed to be accountable in all aspects of the work.
Individuals who did not contribute in all of these ways, but who did participate, are acknowledged below.
We grouped authors into the following four classes of approximately equal contributions and randomly ordered authors within each contribution class.
Drafted multiple sub-sections along with extensive editing, pull request reviews, or discussion: Travers Ching, Brett K. Beaulieu-Jones, Alexandr A. Kalinin, Brian T. Do, Gregory P. Way, Enrico Ferrero, Paul-Michael Agapow, Michael Zietz, Michael M. Hoffman.
Edited the manuscript, reviewed pull requests, and developed Manubot: Daniel S. Himmelstein.
Drafted one or more sub-sections: Wei Xie, Gail L. Rosen, Benjamin J. Lengerich, Johnny Israeli, Jack Lanchantin, Stephen Woloszynek, Anne E. Carpenter, Avanti Shrikumar, Jinbo Xu, Evan M. Cofer, Christopher A. Lavender, Srinivas C. Turaga, Amr M. Alexandari, Zhiyong Lu.
Drafted sub-sections, edited the manuscript, reviewed pull requests, and coordinated co-authors: Anthony Gitter, Casey S. Greene.
Revised specific sub-sections or supervised drafting one or more sub-sections: David J. Harris, Dave DeCaprio, Yanjun Qi, Anshul Kundaje, Yifan Peng, Laura K. Wiley, Marwin H.S. Segler, Simina M. Boca, S. Joshua Swamidass, Austin Huang.
| Author | Competing Interests | Last Reviewed |
|---|---|---|
| Travers Ching | None | 2017-05-26 |
| Daniel S. Himmelstein | None | 2017-05-26 |
| Brett K. Beaulieu-Jones | None | 2017-05-26 |
| Alexandr A. Kalinin | None | 2017-05-26 |
| Brian T. Do | None | 2017-05-26 |
| Gregory P. Way | None | 2017-05-26 |
| Enrico Ferrero | Full-time employee of GlaxoSmithKline. | 2017-05-26 |
| Paul-Michael Agapow | None | 2017-05-26 |
| Michael Zietz | None | 2017-05-26 |
| Michael M. Hoffman | None | 2017-05-26 |
| Wei Xie | None | 2017-05-26 |
| Gail L. Rosen | None | 2017-05-26 |
| Benjamin J. Lengerich | None | 2017-05-26 |
| Johnny Israeli | None | 2017-05-26 |
| Jack Lanchantin | None | 2017-05-26 |
| Stephen Woloszynek | None | 2017-05-26 |
| Anne E. Carpenter | None | 2017-05-26 |
| Avanti Shrikumar | None | 2017-05-26 |
| Jinbo Xu | None | 2017-05-26 |
| Evan M. Cofer | None | 2017-05-26 |
| Christopher A. Lavender | None | 2017-05-26 |
| Srinivas C. Turaga | None | 2017-05-26 |
| Amr M. Alexandari | None | 2017-05-26 |
| Zhiyong Lu | None | 2017-05-26 |
| David J. Harris | None | 2017-05-26 |
| Dave DeCaprio | None | 2017-05-26 |
| Yanjun Qi | None | 2017-05-26 |
| Anshul Kundaje | Advisory Board of Deep Genomics Inc. | 2017-05-26 |
| Yifan Peng | None | 2017-05-26 |
| Laura K. Wiley | None | 2017-05-26 |
| Marwin H.S. Segler | None | 2017-05-26 |
| Simina M. Boca | None | 2017-05-26 |
| S. Joshua Swamidass | None | 2017-05-26 |
| Austin Huang | None | 2017-05-26 |
| Anthony Gitter | None | 2017-05-26 |
| Casey S. Greene | None | 2017-05-26 |
We acknowledge funding from the Gordon and Betty Moore Foundation awards GBMF4552 (C.S.G. and D.S.H.) and GBMF4563 (D.J.H.); the Howard Hughes Medical Institute (S.C.T.); the National Institutes of Health awards DP2GM123485 (A.K.), P30CA051008 (S.M.B.), R01AI116794 (B.K.B.), R01GM089652 (A.E.C.), R01GM089753 (J.X.), R01LM012222 (S.J.S.), R01LM012482 (S.J.S.), R21CA220398 (S.M.B.), T32GM007753 (B.T.D.), T32HG000046 (G.P.W.), and U54AI117924 (A.G.); the National Institutes of Health Intramural Research Program and National Library of Medicine (Y.P. and Z.L.); the National Science Foundation awards 1245632 (G.L.R.), 1531594 (E.M.C.), and 1564955 (J.X.); the Natural Sciences and Engineering Research Council of Canada award RGPIN-2015-3948 (M.M.H.); and the Roy and Diana Vagelos Scholars Program in the Molecular Life Sciences (M.Z.).
We gratefully acknowledge Christof Angermueller, Kumardeep Chaudhary, Gökcen Eraslan, Mikael Huss, Bharath Ramsundar and Xun Zhu for their discussion of the manuscript and reviewed papers on GitHub. We would like to thank Aaron Sheldon, who contributed text but did not formally approve the manuscript. We would like to thank Anna Greene for a careful proofreading of the manuscript in advance of the first submission. We would like to thank Sebastian Raschka for clarifying edits to the abstract and introduction. We would like to thank Robert Gieseke, Ruibang Luo, Stephen Ra, Sourav Singh, and GitHub user snikumbh for correcting typos, formatting, and references.
1. Big Data: Astronomical or Genomical?
Zachary D. Stephens, Skylar Y. Lee, Faraz Faghri, Roy H. Campbell, Chengxiang Zhai, Miles J. Efron, Ravishankar Iyer, Michael C. Schatz, Saurabh Sinha, Gene E. Robinson
PLOS Biology (2015-07-07) https://doi.org/bdzf
DOI: 10.1371/journal.pbio.1002195 · PMID: 26151137 · PMCID: PMC4494865
2. Deep learning
Yann LeCun, Yoshua Bengio, Geoffrey Hinton
Nature (2015-05-27) https://doi.org/bmqp
DOI: 10.1038/nature14539 · PMID: 26017442
3. Searching for exotic particles in high-energy physics with deep learning
P. Baldi, P. Sadowski, D. Whiteson
Nature Communications (2014-07-02) https://doi.org/f6c8jp
DOI: 10.1038/ncomms5308 · PMID: 24986233
4. Deep learning for computational chemistry
Garrett B. Goh, Nathan O. Hodas, Abhinav Vishnu
Journal of Computational Chemistry (2017-06-15) https://doi.org/f959s6
DOI: 10.1002/jcc.24764 · PMID: 28272810
5. Dermatologist-level classification of skin cancer with deep neural networks
Andre Esteva, Brett Kuprel, Roberto A. Novoa, Justin Ko, Susan M. Swetter, Helen M. Blau, Sebastian Thrun
Nature (2017-01-25) https://doi.org/bxwn
DOI: 10.1038/nature21056 · PMID: 28117445
6. Google’s Neural Machine Translation System: Bridging the Gap between Human and Machine Translation
Yonghui Wu, Mike Schuster, Zhifeng Chen, Quoc V. Le, Mohammad Norouzi, Wolfgang Macherey, Maxim Krikun, Yuan Cao, Qin Gao, Klaus Macherey, … Jeffrey Dean
arXiv (2016-10-11) https://arxiv.org/abs/1609.08144
7. A logical calculus of the ideas immanent in nervous activity
Warren S. McCulloch, Walter Pitts
The Bulletin of Mathematical Biophysics (1943-12) https://doi.org/djsbj6
DOI: 10.1007/bf02478259
8. Analysis of a Four-Layer Series-Coupled Perceptron. II
H. D. Block, B. W. Knight, F. Rosenblatt
Reviews of Modern Physics (1962-01-01) https://doi.org/fhx4tr
DOI: 10.1103/revmodphys.34.135
9. Building high-level features using large scale unsupervised learning
Quoc Le, Marc’Aurelio Ranzato, Rajat Monga, Matthieu Devin, Kai Chen, Greg Corrado, Jeff Dean, Andrew Ng
Google Research (2012) https://research.google/pubs/pub38115/
10. HOGWILD!: A Lock-Free Approach to Parallelizing Stochastic Gradient Descent
Feng Niu, Benjamin Recht, Christopher Re, Stephen J. Wright
arXiv (2011-11-14) https://arxiv.org/abs/1106.5730
11. Deep Learning
Ian Goodfellow, Yoshua Bengio, Aaron Courville
(2016) http://www.deeplearningbook.org/
12. Academy of Management: Andrew S. Grove
Andrew S. Grove
(1998-08-09) http://www.intel.com/pressroom/archive/speeches/ag080998.htm
13. Deep learning for regulatory genomics
Yongjin Park, Manolis Kellis
Nature Biotechnology (2015-08-07) https://doi.org/gcgk8b
DOI: 10.1038/nbt.3313 · PMID: 26252139
14. Applications of Deep Learning in Biomedicine
Polina Mamoshina, Armando Vieira, Evgeny Putin, Alex Zhavoronkov
Molecular Pharmaceutics (2016-03-29) https://doi.org/f8mvtj
DOI: 10.1021/acs.molpharmaceut.5b00982 · PMID: 27007977
15. Deep learning for computational biology
Christof Angermueller, Tanel Pärnamaa, Leopold Parts, Oliver Stegle
Molecular Systems Biology (2016-07-29) https://doi.org/f8xtvh
DOI: 10.15252/msb.20156651 · PMID: 27474269 · PMCID: PMC4965871
16. Deep learning in bioinformatics
Seonwoo Min, Byunghan Lee, Sungroh Yoon
Briefings in Bioinformatics (2016-07-29) https://doi.org/gcgk8v
DOI: 10.1093/bib/bbw068 · PMID: 27473064
17. Computer vision for high content screening
Oren Z. Kraus, Brendan J. Frey
Critical Reviews in Biochemistry and Molecular Biology (2016-01-24) https://doi.org/gcgmc2
DOI: 10.3109/10409238.2015.1135868 · PMID: 26806341
18. Deep learning for healthcare: review, opportunities and challenges
Riccardo Miotto, Fei Wang, Shuang Wang, Xiaoqian Jiang, Joel T Dudley
Briefings in Bioinformatics (2018-11) https://doi.org/gcgk8x
DOI: 10.1093/bib/bbx044 · PMID: 28481991 · PMCID: PMC6455466
19. A survey on deep learning in medical image analysis
Geert Litjens, Thijs Kooi, Babak Ehteshami Bejnordi, Arnaud Arindra Adiyoso Setio, Francesco Ciompi, Mohsen Ghafoorian, Jeroen A. W. M. van der Laak, Bram van Ginneken, Clara I. Sánchez
Medical Image Analysis (2017-12) https://doi.org/gcm56p
DOI: 10.1016/j.media.2017.07.005 · PMID: 28778026
20. Deep Learning in Pharmacogenomics: From Gene Regulation to Patient Stratification
Alexandr A. Kalinin, Gerald A. Higgins, Narathip Reamaroon, S. M. Reza Soroushmehr, Ari Allyn-Feuer, Ivo D. Dinov, Kayvan Najarian, Brian D. Athey
arXiv (2018-05-01) https://arxiv.org/abs/1801.08570
DOI: 10.2217/pgs-2018-0008
21. Deep Learning in Drug Discovery
Erik Gawehn, Jan A. Hiss, Gisbert Schneider
Molecular Informatics (2016-01) https://doi.org/f3kg55
DOI: 10.1002/minf.201501008 · PMID: 27491648
22. Virtual Screening: A Challenge for Deep Learning
Javier Pérez-Sianes, Horacio Pérez-Sánchez, Fernando Díaz
Advances in Intelligent Systems and Computing (2016) https://doi.org/gcgk7k
DOI: 10.1007/978-3-319-40126-3_2
23. A renaissance of neural networks in drug discovery
Igor I. Baskin, David Winkler, Igor V. Tetko
Expert Opinion on Drug Discovery (2016-07-04) https://doi.org/gcgk8s
DOI: 10.1080/17460441.2016.1201262 · PMID: 27295548
24. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes
Joel S. Parker, Michael Mullins, Maggie C. U. Cheang, Samuel Leung, David Voduc, Tammi Vickery, Sherri Davies, Christiane Fauron, Xiaping He, Zhiyuan Hu, … Philip S. Bernard
Journal of Clinical Oncology (2009-03-10) https://doi.org/c2688w
DOI: 10.1200/jco.2008.18.1370 · PMID: 19204204 · PMCID: PMC2667820
25. New Strategies for Triple-Negative Breast Cancer–Deciphering the Heterogeneity
I. A. Mayer, V. G. Abramson, B. D. Lehmann, J. A. Pietenpol
Clinical Cancer Research (2014-02-16) https://doi.org/f5tgp5
DOI: 10.1158/1078-0432.ccr-13-0583 · PMID: 24536073 · PMCID: PMC3962777
26. UNSUPERVISED FEATURE CONSTRUCTION AND KNOWLEDGE EXTRACTION FROM GENOME-WIDE ASSAYS OF BREAST CANCER WITH DENOISING AUTOENCODERS
JIE TAN, MATTHEW UNG, CHAO CHENG, CASEY S GREENE
World Scientific Pub Co Pte Lt (2014-11) https://doi.org/gcgmbs
DOI: 10.1142/9789814644730_0014
27. Mitosis Detection in Breast Cancer Histology Images with Deep Neural Networks
Dan C. Cireşan, Alessandro Giusti, Luca M. Gambardella, Jürgen Schmidhuber
Lecture Notes in Computer Science (2013) https://doi.org/gcgk7t
DOI: 10.1007/978-3-642-40763-5_51 · PMID: 24579167
28. End effector target position learning using feedforward with error back-propagation and recurrent neural networks
J. Zurada
Proceedings of 1994 IEEE International Conference on Neural Networks (ICNN’94) (1994) https://doi.org/10.1109/icnn.1994.374637
DOI: 10.1109/icnn.1994.374637
29. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model
Sheng Wang, Siqi Sun, Zhen Li, Renyu Zhang, Jinbo Xu
PLOS Computational Biology (2017-01-05) https://doi.org/f9ktjn
DOI: 10.1371/journal.pcbi.1005324 · PMID: 28056090 · PMCID: PMC5249242
30. A Deep Learning Network Approach to ab initio Protein Secondary Structure Prediction
Matt Spencer, Jesse Eickholt, Jianlin Cheng
IEEE/ACM Transactions on Computational Biology and Bioinformatics (2015-01-01) https://doi.org/gcgmbc
DOI: 10.1109/tcbb.2014.2343960 · PMID: 25750595 · PMCID: PMC4348072
31. Protein Secondary Structure Prediction Using Deep Convolutional Neural Fields
Sheng Wang, Jian Peng, Jianzhu Ma, Jinbo Xu
Scientific Reports (2016-01-11) https://doi.org/f76r5q
DOI: 10.1038/srep18962 · PMID: 26752681 · PMCID: PMC4707437
32. PEDLA: predicting enhancers with a deep learning-based algorithmic framework
Feng Liu, Hao Li, Chao Ren, Xiaochen Bo, Wenjie Shu
bioRxiv (2016-05-18) https://doi.org/gcgk85
DOI: 10.1101/036129
33. Deep Feature Selection: Theory and Application to Identify Enhancers and Promoters
Yifeng Li, Chih-Yu Chen, Wyeth W. Wasserman
Lecture Notes in Computer Science (2015) https://doi.org/gcgk7g
DOI: 10.1007/978-3-319-16706-0_20
34. DEEP: a general computational framework for predicting enhancers
Dimitrios Kleftogiannis, Panos Kalnis, Vladimir B. Bajic
Nucleic Acids Research (2015-01) https://doi.org/gcgk83
DOI: 10.1093/nar/gku1058 · PMID: 25378307 · PMCID: PMC4288148
35. DANN: a deep learning approach for annotating the pathogenicity of genetic variants
Daniel Quang, Yifei Chen, Xiaohui Xie
Bioinformatics (2015-03-01) https://doi.org/f67rhm
DOI: 10.1093/bioinformatics/btu703 · PMID: 25338716 · PMCID: PMC4341060
36. AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery
Izhar Wallach, Michael Dzamba, Abraham Heifets
arXiv (2015-10-13) https://arxiv.org/abs/1510.02855
37. Deep Learning Applications for Predicting Pharmacological Properties of Drugs and Drug Repurposing Using Transcriptomic Data
Alexander Aliper, Sergey Plis, Artem Artemov, Alvaro Ulloa, Polina Mamoshina, Alex Zhavoronkov
Molecular Pharmaceutics (2016-06-08) https://doi.org/gcgk77
DOI: 10.1021/acs.molpharmaceut.6b00248 · PMID: 27200455 · PMCID: PMC4965264
38. Predicting drug-target interactions using restricted Boltzmann machines
Yuhao Wang, Jianyang Zeng
Bioinformatics (2013-07) https://doi.org/gbddzn
DOI: 10.1093/bioinformatics/btt234 · PMID: 23812976 · PMCID: PMC3694663
39. Deep-Learning-Based Drug–Target Interaction Prediction
Ming Wen, Zhimin Zhang, Shaoyu Niu, Haozhi Sha, Ruihan Yang, Yonghuan Yun, Hongmei Lu
Journal of Proteome Research (2017-03-13) https://doi.org/f9ttp4
DOI: 10.1021/acs.jproteome.6b00618 · PMID: 28264154
40. Deep Learning in Medical Image Analysis
Dinggang Shen, Guorong Wu, Heung-Il Suk
Annual Review of Biomedical Engineering (2017-06-21) https://doi.org/gcgmb4
DOI: 10.1146/annurev-bioeng-071516-044442 · PMID: 28301734 · PMCID: PMC5479722
41. Deep Learning and Structured Prediction for the Segmentation of Mass in Mammograms
Neeraj Dhungel, Gustavo Carneiro, Andrew P. Bradley
Lecture Notes in Computer Science (2015) https://doi.org/gcgk7h
DOI: 10.1007/978-3-319-24553-9_74
42. The Automated Learning of Deep Features for Breast Mass Classification from Mammograms
Neeraj Dhungel, Gustavo Carneiro, Andrew P. Bradley
Lecture Notes in Computer Science (2016) https://doi.org/gcgk7q
DOI: 10.1007/978-3-319-46723-8_13
43. Deep Multi-instance Networks with Sparse Label Assignment for Whole Mammogram Classification
Wentao Zhu, Qi Lou, Yeeleng Scott Vang, Xiaohui Xie
bioRxiv (2016-12-20) https://doi.org/gcgk9r
DOI: 10.1101/095794
44. Adversarial Deep Structural Networks for Mammographic Mass Segmentation
Wentao Zhu, Xiaohui Xie
bioRxiv (2016-12-20) https://doi.org/gcgk9q
DOI: 10.1101/095786
45. A deep learning approach for the analysis of masses in mammograms with minimal user intervention
Neeraj Dhungel, Gustavo Carneiro, Andrew P. Bradley
Medical Image Analysis (2017-04) https://doi.org/f932tm
DOI: 10.1016/j.media.2017.01.009 · PMID: 28171807
46. ImageNet Large Scale Visual Recognition Challenge
Olga Russakovsky, Jia Deng, Hao Su, Jonathan Krause, Sanjeev Satheesh, Sean Ma, Zhiheng Huang, Andrej Karpathy, Aditya Khosla, Michael Bernstein, … Li Fei-Fei
International Journal of Computer Vision (2015-04-11) https://doi.org/gcgk7w
DOI: 10.1007/s11263-015-0816-y
47. Convolutional Neural Networks for Diabetic Retinopathy
Harry Pratt, Frans Coenen, Deborah M. Broadbent, Simon P. Harding, Yalin Zheng
Procedia Computer Science (2016) https://doi.org/gcgk75
DOI: 10.1016/j.procs.2016.07.014
48. Improved Automated Detection of Diabetic Retinopathy on a Publicly Available Dataset Through Integration of Deep Learning
Michael David Abràmoff, Yiyue Lou, Ali Erginay, Warren Clarida, Ryan Amelon, James C. Folk, Meindert Niemeijer
Investigative Opthalmology & Visual Science (2016-10-04) https://doi.org/f9mr6r
DOI: 10.1167/iovs.16-19964 · PMID: 27701631
49. Leveraging uncertainty information from deep neural networks for disease detection
Christian Leibig, Vaneeda Allken, Murat Seçkin Ayhan, Philipp Berens, Siegfried Wahl
bioRxiv (2017-10-18) https://doi.org/gcgk9h
DOI: 10.1101/084210
50. Development and Validation of a Deep Learning Algorithm for Detection of Diabetic Retinopathy in Retinal Fundus Photographs
Varun Gulshan, Lily Peng, Marc Coram, Martin C. Stumpe, Derek Wu, Arunachalam Narayanaswamy, Subhashini Venugopalan, Kasumi Widner, Tom Madams, Jorge Cuadros, … Dale R. Webster
JAMA (2016-12-13) https://doi.org/gcgk7d
DOI: 10.1001/jama.2016.17216 · PMID: 27898976
51. Deep Learning Ensembles for Melanoma Recognition in Dermoscopy Images
Noel Codella, Quoc-Bao Nguyen, Sharath Pankanti, David Gutman, Brian Helba, Allan Halpern, John R. Smith
arXiv (2016-10-19) https://arxiv.org/abs/1610.04662
52. Automated Melanoma Recognition in Dermoscopy Images via Very Deep Residual Networks
Lequan Yu, Hao Chen, Qi Dou, Jing Qin, Pheng-Ann Heng
IEEE Transactions on Medical Imaging (2017-04) https://doi.org/gcgmbh
DOI: 10.1109/tmi.2016.2642839 · PMID: 28026754
53. Extraction of skin lesions from non-dermoscopic images for surgical excision of melanoma
M. Hossein Jafari, Ebrahim Nasr-Esfahani, Nader Karimi, S. M. Reza Soroushmehr, Shadrokh Samavi, Kayvan Najarian
International Journal of Computer Assisted Radiology and Surgery (2017-03-24) https://doi.org/gbhjfj
DOI: 10.1007/s11548-017-1567-8 · PMID: 28342106
54. Melanoma detection by analysis of clinical images using convolutional neural network
E. Nasr-Esfahani, S. Samavi, N. Karimi, S. M. R. Soroushmehr, M. H. Jafari, K. Ward, K. Najarian
Institute of Electrical and Electronics Engineers (IEEE) (2016-08) https://doi.org/gcgk97
DOI: 10.1109/embc.2016.7590963 · PMID: 28268581
55. Detection of age-related macular degeneration via deep learning
P. Burlina, D. E. Freund, N. Joshi, Y. Wolfson, N. M. Bressler
Institute of Electrical and Electronics Engineers (IEEE) (2016-04) https://doi.org/gcgmbb
DOI: 10.1109/isbi.2016.7493240
56. Deep learning with non-medical training used for chest pathology identification
Yaniv Bar, Idit Diamant, Lior Wolf, Hayit Greenspan
SPIE-Intl Soc Optical Eng (2015-03-20) https://doi.org/gcgmbm
DOI: 10.1117/12.2083124
57. Deep Convolutional Neural Networks for Computer-Aided Detection: CNN Architectures, Dataset Characteristics and Transfer Learning
Hoo-Chang Shin, Holger R. Roth, Mingchen Gao, Le Lu, Ziyue Xu, Isabella Nogues, Jianhua Yao, Daniel Mollura, Ronald M. Summers
IEEE Transactions on Medical Imaging (2016-05) https://doi.org/gcgmbg
DOI: 10.1109/tmi.2016.2528162 · PMID: 26886976 · PMCID: PMC4890616
58. High-Throughput Classification of Radiographs Using Deep Convolutional Neural Networks
Alvin Rajkomar, Sneha Lingam, Andrew G. Taylor, Michael Blum, John Mongan
Journal of Digital Imaging (2016-10-11) https://doi.org/gcgk7v
DOI: 10.1007/s10278-016-9914-9 · PMID: 27730417 · PMCID: PMC5267603
59. Deep Learning at Chest Radiography: Automated Classification of Pulmonary Tuberculosis by Using Convolutional Neural Networks
Paras Lakhani, Baskaran Sundaram
Radiology (2017-08) https://doi.org/gbp274
DOI: 10.1148/radiol.2017162326 · PMID: 28436741
60. Classification of breast MRI lesions using small-size training sets: comparison of deep learning approaches
Guy Amit, Rami Ben-Ari, Omer Hadad, Einat Monovich, Noa Granot, Sharbell Hashoul
SPIE-Intl Soc Optical Eng (2017-03-03) https://doi.org/gcgmbn
DOI: 10.1117/12.2249981
61. Improving Computer-Aided Detection Using Convolutional Neural Networks and Random View Aggregation
Holger R. Roth, Le Lu, Jiamin Liu, Jianhua Yao, Ari Seff, Kevin Cherry, Lauren Kim, Ronald M. Summers
IEEE Transactions on Medical Imaging (2016-05) https://doi.org/gcgmbf
DOI: 10.1109/tmi.2015.2482920 · PMID: 26441412
62. 3D Deep Learning for Multi-modal Imaging-Guided Survival Time Prediction of Brain Tumor Patients
Dong Nie, Han Zhang, Ehsan Adeli, Luyan Liu, Dinggang Shen
Lecture Notes in Computer Science (2016) https://doi.org/gcgk7r
DOI: 10.1007/978-3-319-46723-8_25 · PMID: 28149967 · PMCID: PMC5278791
63. Large scale deep learning for computer aided detection of mammographic lesions
Thijs Kooi, Geert Litjens, Bram van Ginneken, Albert Gubern-Mérida, Clara I. Sánchez, Ritse Mann, Ard den Heeten, Nico Karssemeijer
Medical Image Analysis (2017-01) https://doi.org/gcgk74
DOI: 10.1016/j.media.2016.07.007 · PMID: 27497072
64. Deep learning as a tool for increased accuracy and efficiency of histopathological diagnosis
Geert Litjens, Clara I. Sánchez, Nadya Timofeeva, Meyke Hermsen, Iris Nagtegaal, Iringo Kovacs, Christina Hulsbergen - van de Kaa, Peter Bult, Bram van Ginneken, Jeroen van der Laak
Scientific Reports (2016-05-23) https://doi.org/f8mqzq
DOI: 10.1038/srep26286 · PMID: 27212078 · PMCID: PMC4876324
65. Deep Learning for Identifying Metastatic Breast Cancer
Dayong Wang, Aditya Khosla, Rishab Gargeya, Humayun Irshad, Andrew H. Beck
arXiv (2016-06-21) https://arxiv.org/abs/1606.05718
66. Deep Convolutional Neural Networks for Breast Cancer Histology Image Analysis
Alexander Rakhlin, Alexey Shvets, Vladimir Iglovikov, Alexandr A. Kalinin
bioRxiv (2018-04-02) https://doi.org/gc3cfc
DOI: 10.1101/259911
67. Deep learning is effective for the classification of OCT images of normal versus Age-related Macular Degeneration
Cecilia S Lee, Doug M Baughman, Aaron Y Lee
bioRxiv (2016-12-14) https://doi.org/gcgk9n
DOI: 10.1101/094276
68. ImageNet Classification with Deep Convolutional Neural Networks
Alex Krizhevsky, Ilya Sutskever, Geoffrey E. Hinton
Proceedings of the 25th International Conference on Neural Information Processing Systems (2012) http://dl.acm.org/citation.cfm?id=2999134.2999257
69. A shared task involving multi-label classification of clinical free text
John P. Pestian, Christopher Brew, Paweł Matykiewicz, D. J. Hovermale, Neil Johnson, K. Bretonnel Cohen, Włodzisław Duch
Association for Computational Linguistics (ACL) (2007) https://doi.org/b4zv3c
DOI: 10.3115/1572392.1572411
70. ChestX-ray8: Hospital-scale Chest X-ray Database and Benchmarks on Weakly-Supervised Classification and Localization of Common Thorax Diseases
Xiaosong Wang, Yifan Peng, Le Lu, Zhiyong Lu, Mohammadhadi Bagheri, Ronald M. Summers
arXiv (2019-02-01) https://arxiv.org/abs/1705.02315
DOI: 10.1109/cvpr.2017.369
71. NegBio: a high-performance tool for negation and uncertainty detection in radiology reports
Yifan Peng, Xiaosong Wang, Le Lu, Mohammadhadi Bagheri, Ronald Summers, Zhiyong Lu
arXiv (2017-12-29) https://arxiv.org/abs/1712.05898
72. Classification evaluation
Jake Lever, Martin Krzywinski, Naomi Altman
Nature Methods (2016-07-28) https://doi.org/gcgk8h
DOI: 10.1038/nmeth.3945
73. NIH Chest X-ray Dataset
NIH Clinical Center
(2017-09-07) https://nihcc.app.box.com/v/ChestXray-NIHCC
74. Pediatric Bone Age Assessment Using Deep Convolutional Neural Networks
Vladimir Iglovikov, Alexander Rakhlin, Alexandr A. Kalinin, Alexey Shvets
bioRxiv (2018-06-20) https://doi.org/gc3cfb
DOI: 10.1101/234120
75. TaggerOne: joint named entity recognition and normalization with semi-Markov Models
Robert Leaman, Zhiyong Lu
Bioinformatics (2016-09-15) https://doi.org/f855dg
DOI: 10.1093/bioinformatics/btw343 · PMID: 27283952 · PMCID: PMC5018376
76. tmVar: a text mining approach for extracting sequence variants in biomedical literature
C.-H. Wei, B. R. Harris, H.-Y. Kao, Z. Lu
Bioinformatics (2013-04-05) https://doi.org/f4zfd9
DOI: 10.1093/bioinformatics/btt156 · PMID: 23564842 · PMCID: PMC3661051
77. DNorm: disease name normalization with pairwise learning to rank
R. Leaman, R. Islamaj Dogan, Z. Lu
Bioinformatics (2013-08-21) https://doi.org/f5gj9n
DOI: 10.1093/bioinformatics/btt474 · PMID: 23969135 · PMCID: PMC3810844
78. Effects of Semantic Features on Machine Learning-Based Drug Name Recognition Systems: Word Embeddings vs. Manually Constructed Dictionaries
Shengyu Liu, Buzhou Tang, Qingcai Chen, Xiaolong Wang
Information (2015-12-11) https://doi.org/gcgnzj
DOI: 10.3390/info6040848
79. Evaluating Word Representation Features in Biomedical Named Entity Recognition Tasks
Buzhou Tang, Hongxin Cao, Xiaolong Wang, Qingcai Chen, Hua Xu
BioMed Research International (2014) https://doi.org/gb76d4
DOI: 10.1155/2014/240403 · PMID: 24729964 · PMCID: PMC3963372
80. Clinical Abbreviation Disambiguation Using Neural Word Embeddings
yonghui wu, Jun Xu, Yaoyun Zhang, Hua Xu
Association for Computational Linguistics (ACL) (2015) https://doi.org/gcgnx7
DOI: 10.18653/v1/w15-3822
81. Exploiting Task-Oriented Resources to Learn Word Embeddings for Clinical Abbreviation Expansion
Yue Liu, Tao Ge, Kusum Mathews, Heng Ji, Deborah McGuinness
Association for Computational Linguistics (ACL) (2015) https://doi.org/gcgnx5
DOI: 10.18653/v1/w15-3810
82. A Comprehensive Benchmark of Kernel Methods to Extract Protein–Protein Interactions from Literature
Domonkos Tikk, Philippe Thomas, Peter Palaga, Jörg Hakenberg, Ulf Leser
PLoS Computational Biology (2010-07-01) https://doi.org/dsnc6j
DOI: 10.1371/journal.pcbi.1000837 · PMID: 20617200 · PMCID: PMC2895635
83. Improving chemical disease relation extraction with rich features and weakly labeled data
Yifan Peng, Chih-Hsuan Wei, Zhiyong Lu
Journal of Cheminformatics (2016-10-07) https://doi.org/gcgnx3
DOI: 10.1186/s13321-016-0165-z · PMID: 28316651 · PMCID: PMC5054544
84. Evaluation of linguistic features useful in extraction of interactions from PubMed; Application to annotating known, high-throughput and predicted interactions in I2D
Yun Niu, David Otasek, Igor Jurisica
Bioinformatics (2010-01-01) https://doi.org/fv4nfs
DOI: 10.1093/bioinformatics/btp602 · PMID: 19850753 · PMCID: PMC2796811
85. Joint Models for Extracting Adverse Drug Events from Biomedical Text
Fei Li, Yue Zhang, Meishan Zhang, Donghong Ji
Proceedings of the Twenty-Fifth International Joint Conference on Artificial Intelligence (2016) http://dl.acm.org/citation.cfm?id=3060832.3061018
ISBN: 978-1-57735-770-4
86. A neural joint model for entity and relation extraction from biomedical text
Fei Li, Meishan Zhang, Guohong Fu, Donghong Ji
BMC Bioinformatics (2017-03-31) https://doi.org/gcgnx2
DOI: 10.1186/s12859-017-1609-9 · PMID: 28359255 · PMCID: PMC5374588
87. Deep learning for extracting protein-protein interactions from biomedical literature
Yifan Peng, Zhiyong Lu
Association for Computational Linguistics (ACL) (2017) https://doi.org/gcgnzc
DOI: 10.18653/v1/w17-2304
88. A Shortest Dependency Path Based Convolutional Neural Network for Protein-Protein Relation Extraction
Lei Hua, Chanqin Quan
BioMed Research International (2016) https://doi.org/f9jkh9
DOI: 10.1155/2016/8479587 · PMID: 27493967 · PMCID: PMC4963603
89. Multichannel Convolutional Neural Network for Biological Relation Extraction
Chanqin Quan, Lei Hua, Xiao Sun, Wenjun Bai
BioMed Research International (2016) https://doi.org/f9kqr3
DOI: 10.1155/2016/1850404 · PMID: 28053977 · PMCID: PMC5174749
90. A general protein-protein interaction extraction architecture based on word representation and feature selection
Zhenchao Jiang, shuang Li, Degen Huang
International Journal of Data Mining and Bioinformatics (2016) https://doi.org/gcgnx4
DOI: 10.1504/ijdmb.2016.074878
91. Chemical-induced disease relation extraction via convolutional neural network
Jinghang Gu, Fuqing Sun, Longhua Qian, Guodong Zhou
Database (2017) https://doi.org/f92754
DOI: 10.1093/database/bax024 · PMID: 28415073 · PMCID: PMC5467558
92. Drug drug interaction extraction from biomedical literature using syntax convolutional neural network
Zhehuan Zhao, Zhihao Yang, Ling Luo, Hongfei Lin, Jian Wang
Bioinformatics (2016-07-27) https://doi.org/f9nsq7
DOI: 10.1093/bioinformatics/btw486 · PMID: 27466626 · PMCID: PMC5181565
93. Extracting Drug-Drug Interactions with Attention CNNs
Masaki Asada, Makoto Miwa, Yutaka Sasaki
Association for Computational Linguistics (ACL) (2017) https://doi.org/gcgnzb
DOI: 10.18653/v1/w17-2302
94. Drug-drug Interaction Extraction via Recurrent Neural Network with Multiple Attention Layers
Zibo Yi, Shasha Li, Jie Yu, Qingbo Wu
arXiv (2017-05-19) https://arxiv.org/abs/1705.03261
95. DUTIR in BioNLP-ST 2016: Utilizing Convolutional Network and Distributed Representation to Extract Complicate Relations
Honglei Li, Jianhai Zhang, Jian Wang, Hongfei Lin, Zhihao Yang
Association for Computational Linguistics (ACL) (2016) https://doi.org/gcgnx9
DOI: 10.18653/v1/w16-3012
96. Deep Learning with Minimal Training Data: TurkuNLP Entry in the BioNLP Shared Task 2016
Farrokh Mehryary, Jari Björne, Sampo Pyysalo, Tapio Salakoski, Filip Ginter
Association for Computational Linguistics (ACL) (2016) https://doi.org/gcgnx8
DOI: 10.18653/v1/w16-3009
97. Using word embedding for bio-event extraction
Chen Li, Runqing Song, Maria Liakata, Andreas Vlachos, Stephanie Seneff, Xiangrong Zhang
Association for Computational Linguistics (ACL) (2015) https://doi.org/gcgnx6
DOI: 10.18653/v1/w15-3814
98. Embedding assisted prediction architecture for event trigger identification
Yifan Nie, Wenge Rong, Yiyuan Zhang, Yuanxin Ouyang, Zhang Xiong
Journal of Bioinformatics and Computational Biology (2015-05-15) https://doi.org/gcgnxz
DOI: 10.1142/s0219720015410012 · PMID: 25669328
99. Biomedical Event Trigger Identification Using Bidirectional Recurrent Neural Network Based Models
Patchigolla VSS Rahul, Sunil Kumar Sahu, Ashish Anand
arXiv (2017-05-29) https://arxiv.org/abs/1705.09516
100. Deep Learning for Biomedical Information Retrieval: Learning Textual Relevance from Click Logs
Sunil Mohan, Nicolas Fiorini, Sun Kim, Zhiyong Lu
Association for Computational Linguistics (ACL) (2017) https://doi.org/gcgnzd
DOI: 10.18653/v1/w17-2328
101. Realizing the full potential of electronic health records: the role of natural language processing
Lucila Ohno-Machado
Journal of the American Medical Informatics Association (2011-09-01) https://doi.org/cx3q3f
DOI: 10.1136/amiajnl-2011-000501 · PMID: 21846784 · PMCID: PMC3168331
102. Machine-learned solutions for three stages of clinical information extraction: the state of the art at i2b2 2010
Berry de Bruijn, Colin Cherry, Svetlana Kiritchenko, Joel Martin, Xiaodan Zhu
Journal of the American Medical Informatics Association (2011-09) https://doi.org/dk7jfw
DOI: 10.1136/amiajnl-2011-000150 · PMID: 21565856 · PMCID: PMC3168309
103. Bidirectional LSTM-CRF for Clinical Concept Extraction
Raghavendra Chalapathy, Ehsan Zare Borzeshi, Massimo Piccardi
arXiv (2016-11-28) https://arxiv.org/abs/1611.08373
104. Multi-task Deep Neural Networks for Automated Extraction of Primary Site and Laterality Information from Cancer Pathology Reports
Hong-Jun Yoon, Arvind Ramanathan, Georgia Tourassi
Advances in Intelligent Systems and Computing (2017) https://doi.org/gcgk7s
DOI: 10.1007/978-3-319-47898-2_21
105. Efficient Estimation of Word Representations in Vector Space
Tomas Mikolov, Kai Chen, Greg Corrado, Jeffrey Dean
arXiv (2013-09-10) https://arxiv.org/abs/1301.3781
106. Exploring the Application of Deep Learning Techniques on Medical Text Corpora
Minarro-Giménez José Antonio, Marín-Alonso Oscar, Samwald Matthias
Studies in Health Technology and Informatics (2014) https://doi.org/gcgnzh
DOI: 10.3233/978-1-61499-432-9-584
107. Medical Semantic Similarity with a Neural Language Model
Lance De Vine, Guido Zuccon, Bevan Koopman, Laurianne Sitbon, Peter Bruza
Association for Computing Machinery (ACM) (2014) https://doi.org/gcgmbx
DOI: 10.1145/2661829.2661974
108. Automatic Diagnosis Coding of Radiology Reports: A Comparison of Deep Learning and Conventional Classification Methods
Sarvnaz Karimi, Xiang Dai, Hamedh Hassanzadeh, Anthony Nguyen
Association for Computational Linguistics (ACL) (2017) https://doi.org/gcgnzg
DOI: 10.18653/v1/w17-2342
109. WHO | International Classification of Diseases, 11th Revision (ICD-11)
WHO
http://www.who.int/classifications/icd/en/
110. Multi-layer Representation Learning for Medical Concepts
Edward Choi, Mohammad Taha Bahadori, Elizabeth Searles, Catherine Coffey, Michael Thompson, James Bost, Javier Tejedor-Sojo, Jimeng Sun
Association for Computing Machinery (ACM) (2016) https://doi.org/gcgmb3
DOI: 10.1145/2939672.2939823
111. Large-Scale Discovery of Disease-Disease and Disease-Gene Associations
Djordje Gligorijevic, Jelena Stojanovic, Nemanja Djuric, Vladan Radosavljevic, Mihajlo Grbovic, Rob J. Kulathinal, Zoran Obradovic
Scientific Reports (2016-08-31) https://doi.org/f8znpb
DOI: 10.1038/srep32404 · PMID: 27578529 · PMCID: PMC5006166
112. Bidirectional RNN for Medical Event Detection in Electronic Health Records
Abhyuday N Jagannatha, Hong Yu
Proceedings of the conference. Association for Computational Linguistics. North American Chapter. Meeting (2016-06) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5119627/
DOI: 10.18653/v1/n16-1056 · PMID: 27885364 · PMCID: PMC5119627
113. Representations of Time Expressions for Temporal Relation Extraction with Convolutional Neural Networks
Chen Lin, Timothy Miller, Dmitriy Dligach, Steven Bethard, Guergana Savova
Association for Computational Linguistics (ACL) (2017) https://doi.org/gcgnzf
DOI: 10.18653/v1/w17-2341
114. Computational Phenotype Discovery Using Unsupervised Feature Learning over Noisy, Sparse, and Irregular Clinical Data
Thomas A. Lasko, Joshua C. Denny, Mia A. Levy
PLoS ONE (2013-06-24) https://doi.org/f49g5g
DOI: 10.1371/journal.pone.0066341 · PMID: 23826094 · PMCID: PMC3691199
115. Semi-supervised learning of the electronic health record for phenotype stratification
Brett K. Beaulieu-Jones, Casey S. Greene
Journal of Biomedical Informatics (2016-12) https://doi.org/gbw7bv
DOI: 10.1016/j.jbi.2016.10.007 · PMID: 27744022
116. Deep Patient: An Unsupervised Representation to Predict the Future of Patients from the Electronic Health Records
Riccardo Miotto, Li Li, Brian A. Kidd, Joel T. Dudley
Scientific Reports (2016-05-17) https://doi.org/f8n6sg
DOI: 10.1038/srep26094 · PMID: 27185194 · PMCID: PMC4869115
117. Doctor AI: Predicting Clinical Events via Recurrent Neural Networks
Edward Choi, Mohammad Taha Bahadori, Andy Schuetz, Walter F. Stewart, Jimeng Sun
arXiv (2016-09-29) https://arxiv.org/abs/1511.05942
118. DeepCare: A Deep Dynamic Memory Model for Predictive Medicine
Trang Pham, Truyen Tran, Dinh Phung, Svetha Venkatesh
arXiv (2017-04-12) https://arxiv.org/abs/1602.00357
119. Deepr: A Convolutional Net for Medical Records
Phuoc Nguyen, Truyen Tran, Nilmini Wickramasinghe, Svetha Venkatesh
IEEE Journal of Biomedical and Health Informatics (2017-01) https://doi.org/10.1109/jbhi.2016.2633963
DOI: 10.1109/jbhi.2016.2633963
120. Multi-task Prediction of Disease Onsets from Longitudinal Lab Tests
Narges Razavian, Jake Marcus, David Sontag
arXiv (2016-09-22) https://arxiv.org/abs/1608.00647
121. Deep Survival Analysis
Rajesh Ranganath, Adler Perotte, Noémie Elhadad, David Blei
arXiv (2016-09-20) https://arxiv.org/abs/1608.02158
122. Comparison of the performance of neural network methods and Cox regression for censored survival data
Anny Xiang, Pablo Lapuerta, Alex Ryutov, Jonathan Buckley, Stanley Azen
Computational Statistics & Data Analysis (2000-08) https://doi.org/bsgrc8
DOI: 10.1016/s0167-9473(99)00098-5
123. DeepSurv: Personalized Treatment Recommender System Using A Cox Proportional Hazards Deep Neural Network
Jared Katzman, Uri Shaham, Jonathan Bates, Alexander Cloninger, Tingting Jiang, Yuval Kluger
arXiv (2018-03-09) https://arxiv.org/abs/1606.00931
DOI: 10.1186/s12874-018-0482-1
124. Deep Exponential Families
Rajesh Ranganath, Linpeng Tang, Laurent Charlin, David M. Blei
arXiv (2014-11-10) https://arxiv.org/abs/1411.2581v1
125. Stochastic Variational Inference
Matt Hoffman, David M. Blei, Chong Wang, John Paisley
arXiv (2013-04-24) https://arxiv.org/abs/1206.7051
126. Hierarchical Variational Models
Rajesh Ranganath, Dustin Tran, David M. Blei
arXiv (2016-06-01) https://arxiv.org/abs/1511.02386
127. A machine learning-based framework to identify type 2 diabetes through electronic health records
Tao Zheng, Wei Xie, Liling Xu, Xiaoying He, Ya Zhang, Mingrong You, Gong Yang, You Chen
International Journal of Medical Informatics (2017-01) https://doi.org/f9hkxb
DOI: 10.1016/j.ijmedinf.2016.09.014 · PMID: 27919371 · PMCID: PMC5144921
128. Implementations by Phenotype | PheKB https://phekb.org/implementations
129. Electronic medical record phenotyping using the anchor and learn framework
Yoni Halpern, Steven Horng, Youngduck Choi, David Sontag
Journal of the American Medical Informatics Association (2016-07) https://doi.org/f84xjd
DOI: 10.1093/jamia/ocw011 · PMID: 27107443 · PMCID: PMC4926745
130. Data Programming: Creating Large Training Sets, Quickly
Alexander Ratner, Christopher De Sa, Sen Wu, Daniel Selsam, Christopher Ré
arXiv (2018-12-10) https://arxiv.org/abs/1605.07723
131. Data is the New Oil
Michael Palmer
ANA Marketing Maestros (2006-11) http://ana.blogs.com/maestros/2006/11/data_is_the_new.html
132. “Data is the New Oil” — A Ludicrous Proposition
Michael Haupt
Medium (2018-04-23) https://medium.com/project-2030/data-is-the-new-oil-a-ludicrous-proposition-1d91bba4f294
133. Data Programming: Machine Learning with Weak Supervision
Alex Ratner, Stephen Bach, Chris Ré
(2016-09-19) http://hazyresearch.github.io/snorkel/blog/weak_supervision.html
134. Mining electronic health records: towards better research applications and clinical care
Peter B. Jensen, Lars J. Jensen, Søren Brunak
Nature Reviews Genetics (2012-05-02) https://doi.org/bdzn
DOI: 10.1038/nrg3208 · PMID: 22549152
135. Methods and dimensions of electronic health record data quality assessment: enabling reuse for clinical research
N. G. Weiskopf, C. Weng
Journal of the American Medical Informatics Association (2013-01-01) https://doi.org/f4mbpb
DOI: 10.1136/amiajnl-2011-000681 · PMID: 22733976 · PMCID: PMC3555312
136. Impact of electronic health record systems on information integrity: quality and safety implications
Sue Bowman
Perspectives in health information management (2013-10-01) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3797550/
PMID: 24159271 · PMCID: PMC3797550
137. Secondary Use of EHR: Data Quality Issues and Informatics Opportunities
Taxiarchis Botsis, Gunnar Hartvigsen, Fei Chen, Chunhua Weng
Summit on translational bioinformatics (2010-03-01) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3041534/
PMID: 21347133 · PMCID: PMC3041534
138. Have DRG-based prospective payment systems influenced the number of secondary diagnoses in health care administrative data?
Lisbeth Serdén, Rikard Lindqvist, Måns Rosén
Health Policy (2003-08) https://doi.org/dpgs86
DOI: 10.1016/s0168-8510(02)00208-7
139. Why Patient Matching Is a Challenge: Research on Master Patient Index (MPI) Data Discrepancies in Key Identifying Fields
Beth Haenke Just, David Marc, Megan Munns, Ryan Sandefer
Perspectives in health information management (2016-04-01) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4832129/
PMID: 27134610 · PMCID: PMC4832129
140. Identifying and mitigating biases in EHR laboratory tests
Rimma Pivovarov, David J. Albers, Jorge L. Sepulveda, Noémie Elhadad
Journal of Biomedical Informatics (2014-10) https://doi.org/f6mgds
DOI: 10.1016/j.jbi.2014.03.016 · PMID: 24727481 · PMCID: PMC4194228
141. Using electronic health records for clinical research: The case of the EHR4CR project
Georges De Moor, Mats Sundgren, Dipak Kalra, Andreas Schmidt, Martin Dugas, Brecht Claerhout, Töresin Karakoyun, Christian Ohmann, Pierre-Yves Lastic, Nadir Ammour, … Pascal Coorevits
Journal of Biomedical Informatics (2015-02) https://doi.org/gcgk73
DOI: 10.1016/j.jbi.2014.10.006 · PMID: 25463966
142. Healthcare Interoperability Standards Compliance Handbook
Frank Oemig, Robert Snelick
Springer International Publishing (2016) https://doi.org/gcgk7m
DOI: 10.1007/978-3-319-44839-8
143. How sample size influences research outcomes
Jorge Faber, Lilian Martins Fonseca
Dental Press Journal of Orthodontics (2014-08) https://doi.org/gcgmcx
DOI: 10.1590/2176-9451.19.4.027-029.ebo · PMID: 25279518 · PMCID: PMC4296634
144. A review of approaches to identifying patient phenotype cohorts using electronic health records
Chaitanya Shivade, Preethi Raghavan, Eric Fosler-Lussier, Peter J Embi, Noemie Elhadad, Stephen B Johnson, Albert M Lai
Journal of the American Medical Informatics Association (2014-03) https://doi.org/f5tsfq
DOI: 10.1136/amiajnl-2013-001935 · PMID: 24201027 · PMCID: PMC3932460
145. STRATEGIES FOR EQUITABLE PHARMACOGENOMIC-GUIDED WARFARIN DOSING AMONG EUROPEAN AND AFRICAN AMERICAN INDIVIDUALS IN A CLINICAL POPULATION
LAURA K. WILEY, JACOB P. VANHOUTEN, DAVID C. SAMUELS, MELINDA C. ALDRICH, DAN M. RODEN, JOSH F. PETERSON, JOSHUA C. DENNY
World Scientific Pub Co Pte Lt (2017-01) https://doi.org/gcgmbr
DOI: 10.1142/9789813207813_0050 · PMID: 27897005 · PMCID: PMC5389380
146. Epidemiological research labelled as a violation of privacy: the case of Estonia
M. Rahu, M. McKee
International Journal of Epidemiology (2008-02-26) https://doi.org/bszgkf
DOI: 10.1093/ije/dyn022 · PMID: 18304955
147. Harnessing next-generation informatics for personalizing medicine: a report from AMIA’s 2014 Health Policy Invitational Meeting
Laura K Wiley, Peter Tarczy-Hornoch, Joshua C Denny, Robert R Freimuth, Casey L Overby, Nigam Shah, Ross D Martin, Indra Neil Sarkar
Journal of the American Medical Informatics Association (2016-03) https://doi.org/f84nww
DOI: 10.1093/jamia/ocv111 · PMID: 26911808 · PMCID: PMC6457095
148. DataSHIELD: taking the analysis to the data, not the data to the analysis
Amadou Gaye, Yannick Marcon, Julia Isaeva, Philippe LaFlamme, Andrew Turner, Elinor M Jones, Joel Minion, Andrew W Boyd, Christopher J Newby, Marja-Liisa Nuotio, … Paul R Burton
International Journal of Epidemiology (2014-12) https://doi.org/gcgk82
DOI: 10.1093/ije/dyu188 · PMID: 25261970 · PMCID: PMC4276062
149. ViPAR: a software platform for the Virtual Pooling and Analysis of Research Data
Kim W Carter, KW Carter, RW Francis, M Bresnahan, M Gissler, TK Grønborg, R Gross, N Gunnes, G Hammond, M Hornig, … Z Yusof
International Journal of Epidemiology (2016-04) https://doi.org/f8pzfm
DOI: 10.1093/ije/dyv193 · PMID: 26452388 · PMCID: PMC4864874
150. Reproducibility of computational workflows is automated using continuous analysis
Brett K Beaulieu-Jones, Casey S Greene
Nature Biotechnology (2017-03-13) https://doi.org/f9ttx6
DOI: 10.1038/nbt.3780 · PMID: 28288103 · PMCID: PMC6103790
151. Stealing Machine Learning Models via Prediction APIs
Florian Tramèr, Fan Zhang, Ari Juels, Michael K. Reiter, Thomas Ristenpart
arXiv (2016-10-04) https://arxiv.org/abs/1609.02943
152. The Algorithmic Foundations of Differential Privacy
Cynthia Dwork, Aaron Roth
Foundations and Trends® in Theoretical Computer Science (2013) https://doi.org/gcgmcw
DOI: 10.1561/0400000042
153. Membership Inference Attacks against Machine Learning Models
Reza Shokri, Marco Stronati, Congzheng Song, Vitaly Shmatikov
arXiv (2017-04-04) https://arxiv.org/abs/1610.05820
154. Enabling Privacy-Preserving GWASs in Heterogeneous Human Populations
Sean Simmons, Cenk Sahinalp, Bonnie Berger
Cell Systems (2016-07) https://doi.org/gcgk72
DOI: 10.1016/j.cels.2016.04.013 · PMID: 27453444 · PMCID: PMC4994706
155. Deep Learning with Differential Privacy
Martin Abadi, Andy Chu, Ian Goodfellow, H. Brendan McMahan, Ilya Mironov, Kunal Talwar, Li Zhang
Association for Computing Machinery (ACM) (2016) https://doi.org/gcrnp3
DOI: 10.1145/2976749.2978318
156. Generating Multi-label Discrete Electronic Health Records using
Generative Adversarial Networks
Edward Choi, Siddharth Biswal, Bradley Malin, Jon Duke, Walter F. Stewart, Jimeng Sun
arXiv (2017-03-19) https://arxiv.org/abs/1703.06490v1
157. Real-valued (Medical) Time Series Generation with Recurrent Conditional
GANs
Cristóbal Esteban, Stephanie L. Hyland, Gunnar Rätsch
arXiv (2017-06-08) https://arxiv.org/abs/1706.02633v1
158. Privacy-preserving generative deep neural networks support clinical data sharing
Brett K. Beaulieu-Jones, Zhiwei Steven Wu, Chris Williams, Ran Lee, Sanjeev P. Bhavnani, James Brian Byrd, Casey S. Greene
bioRxiv (2018-12-20) https://doi.org/gcnzrn
DOI: 10.1101/159756
159. Communication-Efficient Learning of Deep Networks from Decentralized Data
Brendan McMahan, Eider Moore, Daniel Ramage, Seth Hampson, Blaise Aguera y Arcas
Artificial Intelligence and Statistics (2017-04-10) http://proceedings.mlr.press/v54/mcmahan17a.html
160. Practical Secure Aggregation for Privacy Preserving Machine Learning
Keith Bonawitz, Vladimir Ivanov, Ben Kreuter, Antonio Marcedone, H. Brendan McMahan, Sarvar Patel, Daniel Ramage, Aaron Segal, Karn Seth
(2017) https://eprint.iacr.org/2017/281
161. Semi-supervised Knowledge Transfer for Deep Learning from Private Training Data
Nicolas Papernot, Martí, N Abadi, Ú, Lfar Erlingsson, Ian Goodfellow, Kunal Talwar
(2016-11-02) https://openreview.net/forum?id=HkwoSDPgg
162. European Union regulations on algorithmic decision-making and a “right
to explanation”
Bryce Goodman, Seth Flaxman
arXiv (2016-06-28) https://arxiv.org/abs/1606.08813v3
DOI: 10.1609/aimag.v38i3.2741
163. Overcoming the Winner’s Curse: Estimating Penetrance Parameters from Case-Control Data
Sebastian Zöllner, Jonathan K. Pritchard
The American Journal of Human Genetics (2007-04) https://doi.org/fk7jsx
DOI: 10.1086/512821 · PMID: 17357068 · PMCID: PMC1852705
164. Sex bias in neuroscience and biomedical research
Annaliese K. Beery, Irving Zucker
Neuroscience & Biobehavioral Reviews (2011-01) https://doi.org/ff34pz
DOI: 10.1016/j.neubiorev.2010.07.002 · PMID: 20620164 · PMCID: PMC3008499
165. Generalization and Dilution of Association Results from European GWAS in Populations of Non-European Ancestry: The PAGE Study
Christopher S. Carlson, Tara C. Matise, Kari E. North, Christopher A. Haiman, Megan D. Fesinmeyer, Steven Buyske, Fredrick R. Schumacher, Ulrike Peters, Nora Franceschini, Marylyn D. Ritchie, … for the PAGE Consortium
PLoS Biology (2013-09-17) https://doi.org/f6b4xt
DOI: 10.1371/journal.pbio.1001661 · PMID: 24068893 · PMCID: PMC3775722
166. New approaches to population stratification in genome-wide association studies
Alkes L. Price, Noah A. Zaitlen, David Reich, Nick Patterson
Nature Reviews Genetics (2010-06-15) https://doi.org/bw853v
DOI: 10.1038/nrg2813 · PMID: 20548291 · PMCID: PMC2975875
167. Retraction
P. Sebastiani, N. Solovieff, A. Puca, S. W. Hartley, E. Melista, S. Andersen, D. A. Dworkis, J. B. Wilk, R. H. Myers, M. H. Steinberg, … T. T. Perls
Science (2011-07-21) https://doi.org/bn9rxq
DOI: 10.1126/science.333.6041.404-a · PMID: 21778381
168. Leakage in data mining
Shachar Kaufman, Saharon Rosset, Claudia Perlich, Ori Stitelman
ACM Transactions on Knowledge Discovery from Data (2012-12-01) https://doi.org/gcgmbv
DOI: 10.1145/2382577.2382579
169. To predict and serve?
Kristian Lum, William Isaac
Significance (2016-10) https://doi.org/gcgmbk
DOI: 10.1111/j.1740-9713.2016.00960.x
170. Equality of Opportunity in Supervised Learning
Moritz Hardt, Eric Price, Nathan Srebro
arXiv (2016-10-11) https://arxiv.org/abs/1610.02413
171. Fair Algorithms for Infinite and Contextual Bandits
Matthew Joseph, Michael Kearns, Jamie Morgenstern, Seth Neel, Aaron Roth
arXiv (2017-06-30) https://arxiv.org/abs/1610.09559
172. The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective
Syed S Mahmood, Daniel Levy, Ramachandran S Vasan, Thomas J Wang
The Lancet (2014-03) https://doi.org/f2qmmn
DOI: 10.1016/s0140-6736(13)61752-3 · PMID: 24084292 · PMCID: PMC4159698
173. Children of the 90s: Coming of age
Helen Pearson
Nature (2012-04-11) https://doi.org/gcgk79
DOI: 10.1038/484155a · PMID: 22498607
174. Nonparametric Estimation from Incomplete Observations
E. L. Kaplan, Paul Meier
Journal of the American Statistical Association (1958-06) https://doi.org/fscrh2
DOI: 10.2307/2281868
175. Temporal disease trajectories condensed from population-wide registry data covering 6.2 million patients
Anders Boeck Jensen, Pope L. Moseley, Tudor I. Oprea, Sabrina Gade Ellesøe, Robert Eriksson, Henriette Schmock, Peter Bjødstrup Jensen, Lars Juhl Jensen, Søren Brunak
Nature Communications (2014-06-24) https://doi.org/f2zs65
DOI: 10.1038/ncomms5022 · PMID: 24959948 · PMCID: PMC4090719
176. Deepr: A Convolutional Net for Medical Records
Phuoc Nguyen, Truyen Tran, Nilmini Wickramasinghe, Svetha Venkatesh
arXiv (2016-07-27) https://arxiv.org/abs/1607.07519
177. Curiosity Creates Cures: The Value and Impact of Basic Research
NIH
(2012-05) https://www.nigms.nih.gov/Education/Documents/curiosity.pdf
178. Multi-omics integration accurately predicts cellular state in unexplored conditions for Escherichia coli
Minseung Kim, Navneet Rai, Violeta Zorraquino, Ilias Tagkopoulos
Nature Communications (2016-10-07) https://doi.org/gcgk8c
DOI: 10.1038/ncomms13090 · PMID: 27713404 · PMCID: PMC5059772
179. Trans-species learning of cellular signaling systems with bimodal deep belief networks
Lujia Chen, Chunhui Cai, Vicky Chen, Xinghua Lu
Bioinformatics (2015-09-15) https://doi.org/f7sb7n
DOI: 10.1093/bioinformatics/btv315 · PMID: 25995230 · PMCID: PMC4668779
180. Learning structure in gene expression data using deep architectures, with an application to gene clustering
Aman Gupta, Haohan Wang, Madhavi Ganapathiraju
bioRxiv (2015-11-16) https://doi.org/gcgk84
DOI: 10.1101/031906
181. Learning a hierarchical representation of the yeast transcriptomic machinery using an autoencoder model
Lujia Chen, Chunhui Cai, Vicky Chen, Xinghua Lu
BMC Bioinformatics (2016-01-11) https://doi.org/gcgmb8
DOI: 10.1186/s12859-015-0852-1 · PMID: 26818848 · PMCID: PMC4895523
182. ADAGE-Based Integration of Publicly Available Pseudomonas aeruginosa Gene Expression Data with Denoising Autoencoders Illuminates Microbe-Host Interactions
Jie Tan, John H. Hammond, Deborah A. Hogan, Casey S. Greene
mSystems (2016-02-23) https://doi.org/gcgmbq
DOI: 10.1128/msystems.00025-15 · PMID: 27822512 · PMCID: PMC5069748
183. Unsupervised extraction of stable expression signatures from public compendia with eADAGE
Jie Tan, Georgia Doing, Kimberley A. Lewis, Courtney E. Price, Kathleen M. Chen, Kyle C. Cady, Barret Perchuk, Michael T. Laub, Deborah A. Hogan, Casey S. Greene
bioRxiv (2017-04-10) https://doi.org/gcgk9c
DOI: 10.1101/078659
184. Gene expression inference with deep learning
Yifei Chen, Yi Li, Rajiv Narayan, Aravind Subramanian, Xiaohui Xie
Bioinformatics (2016-06-15) https://doi.org/f8vmtt
DOI: 10.1093/bioinformatics/btw074 · PMID: 26873929 · PMCID: PMC4908320
185. DeepChrome: Deep-learning for predicting gene expression from histone modifications
Ritambhara Singh, Jack Lanchantin, Gabriel Robins, Yanjun Qi
arXiv (2016-07-08) https://arxiv.org/abs/1607.02078
186. Attend and Predict: Understanding Gene Regulation by Selective Attention on Chromatin
Ritambhara Singh, Jack Lanchantin, Arshdeep Sekhon, Yanjun Qi
arXiv (2017-11-08) https://arxiv.org/abs/1708.00339
187. Integrative Data Analysis of Multi-Platform Cancer Data with a Multimodal Deep Learning Approach
Muxuan Liang, Zhizhong Li, Ting Chen, Jianyang Zeng
IEEE/ACM Transactions on Computational Biology and Bioinformatics (2015-07-01) https://doi.org/gcgmbd
DOI: 10.1109/tcbb.2014.2377729 · PMID: 26357333
188. DNA methylation and human disease
Keith D. Robertson
Nature Reviews Genetics (2005-08) https://doi.org/bms933
DOI: 10.1038/nrg1655 · PMID: 16136652
189. The Key Role of Epigenetics in Human Disease Prevention and Mitigation
Andrew P. Feinberg
New England Journal of Medicine (2018-04-05) https://doi.org/gdcdrv
DOI: 10.1056/nejmra1402513 · PMID: 29617578
190. Genome-scale DNA methylation maps of pluripotent and differentiated cells
Alexander Meissner, Tarjei S. Mikkelsen, Hongcang Gu, Marius Wernig, Jacob Hanna, Andrey Sivachenko, Xiaolan Zhang, Bradley E. Bernstein, Chad Nusbaum, David B. Jaffe, … Eric S. Lander
Nature (2008-07-06) https://doi.org/cmrp73
DOI: 10.1038/nature07107 · PMID: 18600261 · PMCID: PMC2896277
191. Recurrent Variations in DNA Methylation in Human Pluripotent Stem Cells and Their Differentiated Derivatives
Kristopher L. Nazor, Gulsah Altun, Candace Lynch, Ha Tran, Julie V. Harness, Ileana Slavin, Ibon Garitaonandia, Franz-Josef Müller, Yu-Chieh Wang, Francesca S. Boscolo, … Louise C. Laurent
Cell Stem Cell (2012-05) https://doi.org/f3zqxc
DOI: 10.1016/j.stem.2012.02.013 · PMID: 22560082 · PMCID: PMC3348513
192. Age-Related DNA Methylation Changes in Normal Human Prostate Tissues
B. Kwabi-Addo, W. Chung, L. Shen, M. Ittmann, T. Wheeler, J. Jelinek, J.-P. J. Issa
Clinical Cancer Research (2007-07-01) https://doi.org/cnhhjw
DOI: 10.1158/1078-0432.ccr-07-0085 · PMID: 17606710
193. From The Cover: Epigenetic differences arise during the lifetime of monozygotic twins
M. F. Fraga, E. Ballestar, M. F. Paz, S. Ropero, F. Setien, M. L. Ballestar, D. Heine-Suner, J. C. Cigudosa, M. Urioste, J. Benitez, … M. Esteller
Proceedings of the National Academy of Sciences (2005-07-11) https://doi.org/cbq5b4
DOI: 10.1073/pnas.0500398102 · PMID: 16009939 · PMCID: PMC1174919
194. Aging and Environmental Exposures Alter Tissue-Specific DNA Methylation Dependent upon CpG Island Context
Brock C. Christensen, E. Andres Houseman, Carmen J. Marsit, Shichun Zheng, Margaret R. Wrensch, Joseph L. Wiemels, Heather H. Nelson, Margaret R. Karagas, James F. Padbury, Raphael Bueno, … Karl T. Kelsey
PLoS Genetics (2009-08-14) https://doi.org/d3m62p
DOI: 10.1371/journal.pgen.1000602 · PMID: 19680444 · PMCID: PMC2718614
195. Epigenetic Epidemiology of Common Complex Disease: Prospects for Prediction, Prevention, and Treatment
Caroline L. Relton, George Davey Smith
PLoS Medicine (2010-10-26) https://doi.org/bq3kt8
DOI: 10.1371/journal.pmed.1000356 · PMID: 21048988 · PMCID: PMC2964338
196. Principles and challenges of genome-wide DNA methylation analysis
Peter W. Laird
Nature Reviews Genetics (2010-02-02) https://doi.org/bw4pg6
DOI: 10.1038/nrg2732 · PMID: 20125086
197. Review of processing and analysis methods for DNA methylation array data
CS Wilhelm-Benartzi, DC Koestler, MR Karagas, JM Flanagan, BC Christensen, KT Kelsey, CJ Marsit, EA Houseman, R Brown
British Journal of Cancer (2013-08-27) https://doi.org/gb9qvv
DOI: 10.1038/bjc.2013.496 · PMID: 23982603 · PMCID: PMC3777004
198. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis
Yun Liu, Martin J Aryee, Leonid Padyukov, M Daniele Fallin, Espen Hesselberg, Arni Runarsson, Lovisa Reinius, Nathalie Acevedo, Margaret Taub, Marcus Ronninger, … Andrew P Feinberg
Nature Biotechnology (2013-01-20) https://doi.org/f24vb9
DOI: 10.1038/nbt.2487 · PMID: 23334450 · PMCID: PMC3598632
199. Cell-type deconvolution in epigenome-wide association studies: a review and recommendations
Andrew E Teschendorff, Shijie C Zheng
Epigenomics (2017-05) https://doi.org/f97bw4
DOI: 10.2217/epi-2016-0153 · PMID: 28517979
200. Cell-type deconvolution from DNA methylation: a review of recent applications
Alexander J. Titus, Rachel M. Gallimore, Lucas A. Salas, Brock C. Christensen
Human Molecular Genetics (2017-10-01) https://doi.org/gcqwwg
DOI: 10.1093/hmg/ddx275 · PMID: 28977446 · PMCID: PMC5886462
201. Tracing human stem cell lineage during development using DNA methylation
Lucas A. Salas, John K. Wiencke, Devin C. Koestler, Ze Zhang, Brock C. Christensen, Karl T. Kelsey
Genome Research (2018-09) https://doi.org/gd27pd
DOI: 10.1101/gr.233213.117 · PMID: 30072366 · PMCID: PMC6120629
202. Absence of an embryonic stem cell DNA methylation signature in human cancer
Ze Zhang, John K. Wiencke, Devin C. Koestler, Lucas A. Salas, Brock C. Christensen, Karl T. Kelsey
BMC Cancer (2019-07-19) https://doi.org/gf5k3p
DOI: 10.1186/s12885-019-5932-6 · PMID: 31324166 · PMCID: PMC6642562
203. Obesity accelerates epigenetic aging of human liver
S. Horvath, W. Erhart, M. Brosch, O. Ammerpohl, W. von Schonfels, M. Ahrens, N. Heits, J. T. Bell, P.-C. Tsai, T. D. Spector, … J. Hampe
Proceedings of the National Academy of Sciences (2014-10-13) https://doi.org/f6m897
DOI: 10.1073/pnas.1412759111 · PMID: 25313081 · PMCID: PMC4217403
204. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors
Austin Quach, Morgan E. Levine, Toshiko Tanaka, Ake T. Lu, Brian H. Chen, Luigi Ferrucci, Beate Ritz, Stefania Bandinelli, Marian L. Neuhouser, Jeannette M. Beasley, … Steve Horvath
Aging (2017-02-14) https://doi.org/gf282v
DOI: 10.18632/aging.101168 · PMID: 28198702 · PMCID: PMC5361673
205. DNA methylation age of human tissues and cell types
Steve Horvath
Genome Biology (2013) https://doi.org/pcx
DOI: 10.1186/gb-2013-14-10-r115 · PMID: 24138928 · PMCID: PMC4015143
206. Methylation-Based Biological Age and Breast Cancer Risk
Jacob K Kresovich, Zongli Xu, Katie M O’Brien, Clarice R Weinberg, Dale P Sandler, Jack A Taylor
JNCI: Journal of the National Cancer Institute (2019-10) https://doi.org/gf2nqn
DOI: 10.1093/jnci/djz020 · PMID: 30794318 · PMCID: PMC6792078
207. DNA methylation arrays as surrogate measures of cell mixture distribution
Eugene Andres Houseman, William P Accomando, Devin C Koestler, Brock C Christensen, Carmen J Marsit, Heather H Nelson, John K Wiencke, Karl T Kelsey
BMC Bioinformatics (2012-05-08) https://doi.org/gb8vmz
DOI: 10.1186/1471-2105-13-86 · PMID: 22568884 · PMCID: PMC3532182
208. An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray
Lucas A. Salas, Devin C. Koestler, Rondi A. Butler, Helen M. Hansen, John K. Wiencke, Karl T. Kelsey, Brock C. Christensen
Genome Biology (2018-05-29) https://doi.org/gf2nqq
DOI: 10.1186/s13059-018-1448-7 · PMID: 29843789 · PMCID: PMC5975716
209. Reference-free deconvolution of DNA methylation data and mediation by cell composition effects
E. Andres Houseman, Molly L. Kile, David C. Christiani, Tan A. Ince, Karl T. Kelsey, Carmen J. Marsit
BMC Bioinformatics (2016-06-29) https://doi.org/gf2nqp
DOI: 10.1186/s12859-016-1140-4 · PMID: 27358049 · PMCID: PMC4928286
210. DeepSignal: detecting DNA methylation state from Nanopore sequencing reads using deep-learning
Peng Ni, Neng Huang, Feng Luo, Jianxin Wang
bioRxiv (2018-08-06) https://doi.org/gf2f8t
DOI: 10.1101/385849
211. DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning
Christof Angermueller, Heather J. Lee, Wolf Reik, Oliver Stegle
Genome Biology (2017-04-11) https://doi.org/gcgmcc
DOI: 10.1186/s13059-017-1189-z · PMID: 28395661 · PMCID: PMC5387360
212. MRCNN: a deep learning model for regression of genome-wide DNA methylation
Qi Tian, Jianxiao Zou, Jianxiong Tang, Yuan Fang, Zhongli Yu, Shicai Fan
BMC Genomics (2019-04-04) https://doi.org/gf48g6
DOI: 10.1186/s12864-019-5488-5 · PMID: 30967120 · PMCID: PMC6457069
213. A deep belief network system for prediction of DNA methylation
Mohammed Khwaja, Melpomeni Kalofonou, Chris Toumazou
Institute of Electrical and Electronics Engineers (IEEE) (2017-10) https://doi.org/gf2f8z
DOI: 10.1109/biocas.2017.8325078
214. Predicting DNA Methylation State of CpG Dinucleotide Using Genome Topological Features and Deep Networks
Yiheng Wang, Tong Liu, Dong Xu, Huidong Shi, Chaoyang Zhang, Yin-Yuan Mo, Zheng Wang
Scientific Reports (2016-01-22) https://doi.org/f77m9h
DOI: 10.1038/srep19598 · PMID: 26797014 · PMCID: PMC4726425
215. Predicting DNA methylation states with hybrid information based deep-learning model
Laiyi Fu, Qinke Peng, Ling Chai
IEEE/ACM Transactions on Computational Biology and Bioinformatics (2019) https://doi.org/gf2f82
DOI: 10.1109/tcbb.2019.2909237 · PMID: 30951477
216. E 2 M: A Deep Learning Framework for Associating Combinatorial Methylation Patterns with Gene Expression: Supplementary
Jianhao Peng, Idoia Ochoa, Olgica Milenkovic
bioRxiv (2019-01-22) https://doi.org/gf2hjm
DOI: 10.1101/527044
217. Predicting Methylation from Sequence and Gene Expression Using Deep Learning with Attention
Alona Levy-Jurgenson, Xavier Tekpli, Vessela N. Kristensen, Zohar Yakhini
bioRxiv (2018-12-09) https://doi.org/gf2f8x
DOI: 10.1101/491357
218. D-GPM: a deep learning method for gene promoter methylation inference
Xingxin Pan, Biao Liu, Xingzhao Wen, Yulu Liu, Xiuqing Zhang, Shengbin Li, Shuaicheng Li
bioRxiv (2018-11-24) https://doi.org/gfh798
DOI: 10.1101/438218
219. Deep Recurrent Attention Models for Histopathological Image Analysis
Alexandre Momeni, Marc Thibault, Olivier Gevaert
bioRxiv (2018-10-14) https://doi.org/gf2f8w
DOI: 10.1101/438341
220. Residual Deep Convolutional Neural Network Predicts MGMT Methylation Status
Panagiotis Korfiatis, Timothy L. Kline, Daniel H. Lachance, Ian F. Parney, Jan C. Buckner, Bradley J. Erickson
Journal of Digital Imaging (2017-08-07) https://doi.org/gf2f8q
DOI: 10.1007/s10278-017-0009-z · PMID: 28785873 · PMCID: PMC5603430
221. A deep learning framework for imputing missing values in genomic data
Yeping Lina Qiu, Hong Zheng, Olivier Gevaert
bioRxiv (2018-09-03) https://doi.org/gf2f8v
DOI: 10.1101/406066
222. A deep neural network based regression model for triglyceride concentrations prediction using epigenome-wide DNA methylation profiles
Md. Mohaiminul Islam, Ye Tian, Yan Cheng, Yang Wang, Pingzhao Hu
BMC Proceedings (2018-09-17) https://doi.org/gf2f84
DOI: 10.1186/s12919-018-0121-1 · PMID: 30263040 · PMCID: PMC6157031
223. Data mining and machine learning approaches for the integration of genome-wide association and methylation data: methodology and main conclusions from GAW20
Burcu Darst, Corinne D. Engelman, Ye Tian, Justo Lorenzo Bermejo
BMC Genetics (2018-09-17) https://doi.org/gf2f83
DOI: 10.1186/s12863-018-0646-3 · PMID: 30255774 · PMCID: PMC6157271
224. Convolutional Neural Networks In Classifying Cancer Through DNA Methylation
Soham Chatterjee, Archana Iyer, Satya Avva, Abhai Kollara, Malaikannan Sankarasubbu
arXiv (2018-07-26) https://arxiv.org/abs/1807.09617
225. A Deep Autoencoder System for Differentiation of Cancer Types Based on DNA Methylation State
Mohammed Khwaja, Melpomeni Kalofonou, Chris Toumazou
arXiv (2018-10-08) https://arxiv.org/abs/1810.01243
226. MethylNet: An Automated and Modular Deep Learning Approach for DNA Methylation Analysis
Joshua J Levy, Alexander J Titus, Curtis L Petersen, David Chen, Lucas A Salas, Brock C Christensen
bioRxiv (2019-12-17) https://doi.org/gf48g5
DOI: 10.1101/692665
227. Visualizing Data using t-SNE
Laurens van der Maaten, Geoffrey Hinton
Journal of Machine Learning Research (2008) http://www.jmlr.org/papers/v9/vandermaaten08a.html
228. Deep Neural Network for Analysis of DNA Methylation Data
Hong Yu, Zhanyu Ma
arXiv (2020-01-29) https://arxiv.org/abs/1808.01359
229. A New Dimension of Breast Cancer Epigenetics - Applications of Variational Autoencoders with DNA Methylation
Alexander J. Titus, Carly A. Bobak, Brock C. Christensen
Scitepress (2018) https://doi.org/gd58sg
DOI: 10.5220/0006636401400145
230. Extracting a biologically relevant latent space from cancer transcriptomes with variational autoencoders
Gregory P. Way, Casey S. Greene
World Scientific Pub Co Pte Lt (2018-01) https://doi.org/gfspsd
DOI: 10.1142/9789813235533_0008
231. Unsupervised deep learning with variational autoencoders applied to breast tumor genome-wide DNA methylation data with biologic feature extraction
Alexander J. Titus, Owen M. Wilkins, Carly A. Bobak, Brock C. Christensen
bioRxiv (2018-11-07) https://doi.org/gf2hfb
DOI: 10.1101/433763
232. Exploring DNA Methylation Data of Lung Cancer Samples with Variational Autoencoders
Zhenxing Wang, Yadong Wang
Institute of Electrical and Electronics Engineers (IEEE) (2018-12) https://doi.org/gf2hff
DOI: 10.1109/bibm.2018.8621365
233. Parameter tuning is a key part of dimensionality reduction via deep variational autoencoders for single cell RNA transcriptomics
Qiwen Hu, Casey S. Greene
bioRxiv (2018-09-20) https://doi.org/gdxxjf
DOI: 10.1101/385534
234. RNA mis-splicing in disease
Marina M. Scotti, Maurice S. Swanson
Nature Reviews Genetics (2015-11-23) https://doi.org/gcgk8m
DOI: 10.1038/nrg.2015.3 · PMID: 26593421 · PMCID: PMC5993438
235. RNA splicing is a primary link between genetic variation and disease
Yang I. Li, Bryce van de Geijn, Anil Raj, David A. Knowles, Allegra A. Petti, David Golan, Yoav Gilad, Jonathan K. Pritchard
Science (2016-04-29) https://doi.org/f8j95d
DOI: 10.1126/science.aad9417 · PMID: 27126046 · PMCID: PMC5182069
236. Deciphering the splicing code
Yoseph Barash, John A. Calarco, Weijun Gao, Qun Pan, Xinchen Wang, Ofer Shai, Benjamin J. Blencowe, Brendan J. Frey
Nature (2010-05) https://doi.org/cvk585
DOI: 10.1038/nature09000 · PMID: 20445623
237. Bayesian prediction of tissue-regulated splicing using RNA sequence and cellular context
Hui Yuan Xiong, Yoseph Barash, Brendan J. Frey
Bioinformatics (2011-09-15) https://doi.org/c9jnt2
DOI: 10.1093/bioinformatics/btr444 · PMID: 21803804
238. The human splicing code reveals new insights into the genetic determinants of disease
H. Y. Xiong, B. Alipanahi, L. J. Lee, H. Bretschneider, D. Merico, R. K. C. Yuen, Y. Hua, S. Gueroussov, H. S. Najafabadi, T. R. Hughes, … B. J. Frey
Science (2014-12-18) https://doi.org/f6wzj2
DOI: 10.1126/science.1254806 · PMID: 25525159 · PMCID: PMC4362528
239. Integrative Deep Models for Alternative Splicing
Anupama Jha, Matthew R. Gazzara, Yoseph Barash
bioRxiv (2017-01-31) https://doi.org/gcgk9v
DOI: 10.1101/104869
240. Imputation for transcription factor binding predictions based on deep learning
Qian Qin, Jianxing Feng
PLOS Computational Biology (2017-02-24) https://doi.org/f9rgsc
DOI: 10.1371/journal.pcbi.1005403 · PMID: 28234893 · PMCID: PMC5345877
241. Learning the Sequence Determinants of Alternative Splicing from Millions of Random Sequences
Alexander B. Rosenberg, Rupali P. Patwardhan, Jay Shendure, Georg Seelig
Cell (2015-10) https://doi.org/f7x2wb
DOI: 10.1016/j.cell.2015.09.054 · PMID: 26496609
242. MECHANISMS IN ENDOCRINOLOGY: Alternative splicing: the new frontier in diabetes research
Jonàs Juan-Mateu, Olatz Villate, Décio L Eizirik
European Journal of Endocrinology (2016-05) https://doi.org/f8nccm
DOI: 10.1530/eje-15-0916 · PMID: 26628584 · PMCID: PMC5331159
243. Absence of a simple code: how transcription factors read the genome
Matthew Slattery, Tianyin Zhou, Lin Yang, Ana Carolina Dantas Machado, Raluca Gordân, Remo Rohs
Trends in Biochemical Sciences (2014-09) https://doi.org/xrn
DOI: 10.1016/j.tibs.2014.07.002 · PMID: 25129887 · PMCID: PMC4149858
244. An integrated encyclopedia of DNA elements in the human genome
The ENCODE Project Consortium
Nature (2012-09-05) https://doi.org/bg9d
DOI: 10.1038/nature11247 · PMID: 22955616 · PMCID: PMC3439153
245. DNA binding sites: representation and discovery
G. D. Stormo
Bioinformatics (2000-01-01) https://doi.org/bdqmmh
DOI: 10.1093/bioinformatics/16.1.16 · PMID: 10812473
246. MEME SUITE: tools for motif discovery and searching
T. L. Bailey, M. Boden, F. A. Buske, M. Frith, C. E. Grant, L. Clementi, J. Ren, W. W. Li, W. S. Noble
Nucleic Acids Research (2009-05-20) https://doi.org/bzkmbr
DOI: 10.1093/nar/gkp335 · PMID: 19458158 · PMCID: PMC2703892
247. Evaluation of methods for modeling transcription factor sequence specificity
Matthew T Weirauch, Atina Cote, Raquel Norel, Matti Annala, Yue Zhao, Todd R Riley, Julio Saez-Rodriguez, Thomas Cokelaer, Anastasia Vedenko, Shaheynoor Talukder, … DREAM5 Consortium
Nature Biotechnology (2013-01-27) https://doi.org/f4pprj
DOI: 10.1038/nbt.2486 · PMID: 23354101 · PMCID: PMC3687085
248. High Resolution Models of Transcription Factor-DNA Affinities Improve In Vitro and In Vivo Binding Predictions
Phaedra Agius, Aaron Arvey, William Chang, William Stafford Noble, Christina Leslie
PLoS Computational Biology (2010-09-09) https://doi.org/c8n9s7
DOI: 10.1371/journal.pcbi.1000916 · PMID: 20838582 · PMCID: PMC2936517
249. Enhanced Regulatory Sequence Prediction Using Gapped k-mer Features
Mahmoud Ghandi, Dongwon Lee, Morteza Mohammad-Noori, Michael A. Beer
PLoS Computational Biology (2014-07-17) https://doi.org/gcgmcf
DOI: 10.1371/journal.pcbi.1003711 · PMID: 25033408 · PMCID: PMC4102394
250. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning
Babak Alipanahi, Andrew Delong, Matthew T Weirauch, Brendan J Frey
Nature Biotechnology (2015-07-27) https://doi.org/f7mkrd
DOI: 10.1038/nbt.3300 · PMID: 26213851
251. RNA-protein binding motifs mining with a new hybrid deep learning based cross-domain knowledge integration approach
Xiaoyong Pan, Hong-Bin Shen
BMC Bioinformatics (2017-02-28) https://doi.org/gcgmb9
DOI: 10.1186/s12859-017-1561-8 · PMID: 28245811 · PMCID: PMC5331642
252. Convolutional neural network architectures for predicting DNA–protein binding
Haoyang Zeng, Matthew D. Edwards, Ge Liu, David K. Gifford
Bioinformatics (2016-06-15) https://doi.org/gcgk8z
DOI: 10.1093/bioinformatics/btw255 · PMID: 27307608 · PMCID: PMC4908339
253. Deep Motif Dashboard: Visualizing and Understanding Genomic Sequences Using Deep Neural Networks
Jack Lanchantin, Ritambhara Singh, Beilun Wang, Yanjun Qi
arXiv (2016-10-20) https://arxiv.org/abs/1608.03644
254. Convolutional Kitchen Sinks for Transcription Factor Binding Site Prediction
Alyssa Morrow, Vaishaal Shankar, Devin Petersohn, Anthony Joseph, Benjamin Recht, Nir Yosef
arXiv (2017-06-02) https://arxiv.org/abs/1706.00125
255. Biological Sequence Modeling with Convolutional Kernel Networks
Dexiong Chen, Laurent Jacob, Julien Mairal
bioRxiv (2019-01-29) https://doi.org/gcsmgj
DOI: 10.1101/217257
256. Reverse-complement parameter sharing improves deep learning models for genomics
Avanti Shrikumar, Peyton Greenside, Anshul Kundaje
bioRxiv (2017-01-27) https://doi.org/gcgk9t
DOI: 10.1101/103663
257. Separable Fully Connected Layers Improve Deep Learning Models For Genomics
Amr Mohamed Alexandari, Avanti Shrikumar, Anshul Kundaje
bioRxiv (2017-07-07) https://doi.org/gcsmgh
DOI: 10.1101/146431
258. Predicting effects of noncoding variants with deep learning–based sequence model
Jian Zhou, Olga G Troyanskaya
Nature Methods (2015-08-24) https://doi.org/gcgk8g
DOI: 10.1038/nmeth.3547 · PMID: 26301843 · PMCID: PMC4768299
259. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences
Daniel Quang, Xiaohui Xie
Nucleic Acids Research (2016-06-20) https://doi.org/f8v4wj
DOI: 10.1093/nar/gkw226 · PMID: 27084946 · PMCID: PMC4914104
260. Sequence and chromatin determinants of cell-type-specific transcription factor binding
A. Arvey, P. Agius, W. S. Noble, C. Leslie
Genome Research (2012-09-05) https://doi.org/f37s7h
DOI: 10.1101/gr.127712.111 · PMID: 22955984 · PMCID: PMC3431489
261. Analysis of computational footprinting methods for DNase sequencing experiments
Eduardo G Gusmao, Manuel Allhoff, Martin Zenke, Ivan G Costa
Nature Methods (2016-02-22) https://doi.org/f8hz8z
DOI: 10.1038/nmeth.3772 · PMID: 26901649
262. ENCODE-DREAM in vivo Transcription Factor Binding Site Prediction Challenge (2017) https://www.synapse.org/#!Synapse:syn6131484/wiki/402026
263. FactorNet: a deep learning framework for predicting cell type specific transcription factor binding from nucleotide-resolution sequential data
Daniel Quang, Xiaohui Xie
bioRxiv (2017-06-28) https://doi.org/gcpvb3
DOI: 10.1101/151274
264. Learning from mistakes: Accurate prediction of cell type-specific transcription factor binding
Jens Keilwagen, Stefan Posch, Jan Grau
bioRxiv (2018-06-12) https://doi.org/gcsmgk
DOI: 10.1101/230011
265. Transfer String Kernel for Cross-Context DNA-Protein Binding Prediction
Ritambhara Singh, Jack Lanchantin, Gabriel Robins, Yanjun Qi
IEEE/ACM Transactions on Computational Biology and Bioinformatics (2019-09-01) https://doi.org/gcsmhp
DOI: 10.1109/tcbb.2016.2609918 · PMID: 27654939
266. Learning Transferable Features with Deep Adaptation Networks
Mingsheng Long, Yue Cao, Jianmin Wang, Michael I. Jordan
arXiv (2015-05-28) https://arxiv.org/abs/1502.02791
267. Domain-Adversarial Training of Neural Networks
Yaroslav Ganin, Evgeniya Ustinova, Hana Ajakan, Pascal Germain, Hugo Larochelle, François Laviolette, Mario Marchand, Victor Lempitsky
arXiv (2016-05-27) https://arxiv.org/abs/1505.07818
268. Learning Important Features Through Propagating Activation Differences
Avanti Shrikumar, Peyton Greenside, Anshul Kundaje
arXiv (2019-10-15) https://arxiv.org/abs/1704.02685
269. The state of the art of mammalian promoter recognition
T. Werner
Briefings in Bioinformatics (2003-01-01) https://doi.org/cz8869
DOI: 10.1093/bib/4.1.22 · PMID: 12715831
270. Detection of RNA polymerase II promoters and polyadenylation sites in human DNA sequence
Sherri Matis, Ying Xu, Manesh Shah, Xiaojun Guan, J.Ralph Einstein, Richard Mural, Edward Uberbacher
Computers & Chemistry (1996-03) https://doi.org/b3f4ch
DOI: 10.1016/s0097-8485(96)80015-5
271. Recognition of prokaryotic and eukaryotic promoters using convolutional deep learning neural networks
Ramzan Kh. Umarov, Victor V. Solovyev
PLOS ONE (2017-02-03) https://doi.org/gcgmcr
DOI: 10.1371/journal.pone.0171410 · PMID: 28158264 · PMCID: PMC5291440
272. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage
T. Shiraki, S. Kondo, S. Katayama, K. Waki, T. Kasukawa, H. Kawaji, R. Kodzius, A. Watahiki, M. Nakamura, T. Arakawa, … Y. Hayashizaki
Proceedings of the National Academy of Sciences (2003-12-08) https://doi.org/c8d26z
DOI: 10.1073/pnas.2136655100 · PMID: 14663149 · PMCID: PMC307644
273. Genome-wide characterization of transcriptional start sites in humans by integrative transcriptome analysis
R. Yamashita, N. P. Sathira, A. Kanai, K. Tanimoto, T. Arauchi, Y. Tanaka, S.-i. Hashimoto, S. Sugano, K. Nakai, Y. Suzuki
Genome Research (2011-03-03) https://doi.org/cf7nzg
DOI: 10.1101/gr.110254.110 · PMID: 21372179 · PMCID: PMC3083095
274. Enhancers: five essential questions
Len A. Pennacchio, Wendy Bickmore, Ann Dean, Marcelo A. Nobrega, Gill Bejerano
Nature Reviews Genetics (2013-03-18) https://doi.org/gcgk8n
DOI: 10.1038/nrg3458 · PMID: 23503198 · PMCID: PMC4445073
275. A unified architecture of transcriptional regulatory elements
Robin Andersson, Albin Sandelin, Charles G. Danko
Trends in Genetics (2015-08) https://doi.org/f7nnvh
DOI: 10.1016/j.tig.2015.05.007 · PMID: 26073855
276. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks
David R. Kelley, Jasper Snoek, John L. Rinn
Genome Research (2016-07) https://doi.org/f8sw35
DOI: 10.1101/gr.200535.115 · PMID: 27197224 · PMCID: PMC4937568
277. DeepEnhancer: Predicting enhancers by convolutional neural networks
Xu Min, Ning Chen, Ting Chen, Rui Jiang
Institute of Electrical and Electronics Engineers (IEEE) (2016-12) https://doi.org/gcgk96
DOI: 10.1109/bibm.2016.7822593
278. Genome-Wide Prediction of cis-Regulatory Regions Using Supervised Deep Learning Methods
Yifeng Li, Wenqiang Shi, Wyeth W Wasserman
bioRxiv (2016-02-28) https://doi.org/gcgk86
DOI: 10.1101/041616
279. Predicting Enhancer-Promoter Interaction from Genomic Sequence with Deep Neural Networks
Shashank Singh, Yang Yang, Barnabás Póczos, Jian Ma
bioRxiv (2018-02-05) https://doi.org/gcgk9k
DOI: 10.1101/085241
280. A network-biology perspective of microRNA function and dysfunction in cancer
Cameron P. Bracken, Hamish S. Scott, Gregory J. Goodall
Nature Reviews Genetics (2016-10-31) https://doi.org/f9bt2c
DOI: 10.1038/nrg.2016.134 · PMID: 27795564
281. Evolution of microRNA diversity and regulation in animals
Eugene Berezikov
Nature Reviews Genetics (2011-11-18) https://doi.org/dk6596
DOI: 10.1038/nrg3079 · PMID: 22094948
282. Predicting effective microRNA target sites in mammalian mRNAs
Vikram Agarwal, George W Bell, Jin-Wu Nam, David P Bartel
eLife (2015-08-12) https://doi.org/gcgmc6
DOI: 10.7554/elife.05005 · PMID: 26267216 · PMCID: PMC4532895
283. deepTarget: End-to-end Learning Framework for microRNA Target Prediction
using Deep Recurrent Neural Networks
Byunghan Lee, Junghwan Baek, Seunghyun Park, Sungroh Yoon
arXiv (2016-03-30) https://arxiv.org/abs/1603.09123v2
284. deepMiRGene: Deep Neural Network based Precursor microRNA Prediction
Seunghyun Park, Seonwoo Min, Hyunsoo Choi, Sungroh Yoon
arXiv (2016-05-04) https://arxiv.org/abs/1605.00017
285. AUC-Maximized Deep Convolutional Neural Fields for Protein Sequence Labeling
Sheng Wang, Siqi Sun, Jinbo Xu
Lecture Notes in Computer Science (2016) https://doi.org/gcgk7n
DOI: 10.1007/978-3-319-46227-1_1 · PMID: 28884168 · PMCID: PMC5584645
286. MetaPSICOV: combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins
David T. Jones, Tanya Singh, Tomasz Kosciolek, Stuart Tetchner
Bioinformatics (2015-04-01) https://doi.org/f67gz4
DOI: 10.1093/bioinformatics/btu791 · PMID: 25431331 · PMCID: PMC4382908
287. Identification of direct residue contacts in protein-protein interaction by message passing
M. Weigt, R. A. White, H. Szurmant, J. A. Hoch, T. Hwa
Proceedings of the National Academy of Sciences (2008-12-30) https://doi.org/dx4sww
DOI: 10.1073/pnas.0805923106 · PMID: 19116270 · PMCID: PMC2629192
288. Protein 3D Structure Computed from Evolutionary Sequence Variation
Debora S. Marks, Lucy J. Colwell, Robert Sheridan, Thomas A. Hopf, Andrea Pagnani, Riccardo Zecchina, Chris Sander
PLoS ONE (2011-12-07) https://doi.org/cmnkx6
DOI: 10.1371/journal.pone.0028766 · PMID: 22163331 · PMCID: PMC3233603
289. A Unified Multitask Architecture for Predicting Local Protein Properties
Yanjun Qi, Merja Oja, Jason Weston, William Stafford Noble
PLoS ONE (2012-03-26) https://doi.org/gcgmck
DOI: 10.1371/journal.pone.0032235 · PMID: 22461885 · PMCID: PMC3312883
290. Improving prediction of secondary structure, local backbone angles and solvent accessible surface area of proteins by iterative deep learning
Rhys Heffernan, Kuldip Paliwal, James Lyons, Abdollah Dehzangi, Alok Sharma, Jihua Wang, Abdul Sattar, Yuedong Yang, Yaoqi Zhou
Scientific Reports (2015-06-22) https://doi.org/gcgk8q
DOI: 10.1038/srep11476 · PMID: 26098304 · PMCID: PMC4476419
291. Protein secondary structure prediction based on position-specific scoring matrices 1 1Edited by G. Von Heijne
David T Jones
Journal of Molecular Biology (1999-09) https://doi.org/d3fxv7
DOI: 10.1006/jmbi.1999.3091 · PMID: 10493868
292. Deep Supervised and Convolutional Generative Stochastic Network for Protein Secondary Structure Prediction
Jian Zhou, Olga G. Troyanskaya
arXiv (2014-03-07) https://arxiv.org/abs/1403.1347
293. Protein contact prediction by integrating joint evolutionary coupling analysis and supervised learning
Jianzhu Ma, Sheng Wang, Zhiyong Wang, Jinbo Xu
Bioinformatics (2015-11-01) https://doi.org/f7zggf
DOI: 10.1093/bioinformatics/btv472 · PMID: 26275894 · PMCID: PMC4838177
294. Deep architectures for protein contact map prediction
Pietro Di Lena, Ken Nagata, Pierre Baldi
Bioinformatics (2012-10-01) https://doi.org/f4bwr4
DOI: 10.1093/bioinformatics/bts475 · PMID: 22847931 · PMCID: PMC3463120
295. Predicting protein residue–residue contacts using deep networks and boosting
Jesse Eickholt, Jianlin Cheng
Bioinformatics (2012-12) https://doi.org/f4hqfh
DOI: 10.1093/bioinformatics/bts598 · PMID: 23047561 · PMCID: PMC3509494
296. Improved Contact Predictions Using the Recognition of Protein Like Contact Patterns
Marcin J. Skwark, Daniele Raimondi, Mirco Michel, Arne Elofsson
PLoS Computational Biology (2014-11-06) https://doi.org/f25c62
DOI: 10.1371/journal.pcbi.1003889 · PMID: 25375897 · PMCID: PMC4222596
297. RR Results - CASP12 (2017) http://www.predictioncenter.org/casp12/rrc_avrg_results.cgi
298. CAMEO - Continuous Automated Model Evaluation (2017) https://www.cameo3d.org/
299. Predicting membrane protein contacts from non-membrane proteins by deep transfer learning
Zhen Li, Sheng Wang, Yizhou Yu, Jinbo Xu
arXiv (2017-05-11) https://arxiv.org/abs/1704.07207
300. End-to-end differentiable learning of protein structure
Mohammed AlQuraishi
bioRxiv (2018-08-29) https://doi.org/gc3gsf
DOI: 10.1101/265231
301. Single-Particle Cryo-EM at Crystallographic Resolution
Yifan Cheng
Cell (2015-04) https://doi.org/gcnz3b
DOI: 10.1016/j.cell.2015.03.049 · PMID: 25910205 · PMCID: PMC4409662
302. A Primer to Single-Particle Cryo-Electron Microscopy
Yifan Cheng, Nikolaus Grigorieff, Pawel A. Penczek, Thomas Walz
Cell (2015-04) https://doi.org/f692sc
DOI: 10.1016/j.cell.2015.03.050 · PMID: 25910204 · PMCID: PMC4409659
303. SwarmPS: Rapid, semi-automated single particle selection software
David Woolford, Geoffery Ericksson, Rosalba Rothnagel, David Muller, Michael J. Landsberg, Radosav S. Pantelic, Alasdair McDowall, Bernard Pailthorpe, Paul R. Young, Ben Hankamer, Jasmine Banks
Journal of Structural Biology (2007-01) https://doi.org/cqvpns
DOI: 10.1016/j.jsb.2006.04.006 · PMID: 16774837
304. Semi-automated selection of cryo-EM particles in RELION-1.3
Sjors H. W. Scheres
Journal of Structural Biology (2015-02) https://doi.org/f6xkc7
DOI: 10.1016/j.jsb.2014.11.010 · PMID: 25486611 · PMCID: PMC4318617
305. DeepPicker: A deep learning approach for fully automated particle picking in cryo-EM
Feng Wang, Huichao Gong, Gaochao Liu, Meijing Li, Chuangye Yan, Tian Xia, Xueming Li, Jianyang Zeng
Journal of Structural Biology (2016-09) https://doi.org/f8xr4n
DOI: 10.1016/j.jsb.2016.07.006 · PMID: 27424268
306. A deep convolutional neural network approach to single-particle recognition in cryo-electron microscopy
Yanan Zhu, Qi Ouyang, Youdong Mao
BMC Bioinformatics (2017-07-21) https://doi.org/gcnz3c
DOI: 10.1186/s12859-017-1757-y · PMID: 28732461 · PMCID: PMC5521087
307. Massively parallel unsupervised single-particle cryo-EM data clustering via statistical manifold learning
Jiayi Wu, Yong-Bei Ma, Charles Congdon, Bevin Brett, Shuobing Chen, Yaofang Xu, Qi Ouyang, Youdong Mao
PLOS ONE (2017-08-07) https://doi.org/gbqwkp
DOI: 10.1371/journal.pone.0182130 · PMID: 28786986 · PMCID: PMC5546606
308. Protein–Protein Interactions Essentials: Key Concepts to Building and Analyzing Interactome Networks
Javier De Las Rivas, Celia Fontanillo
PLoS Computational Biology (2010-06-24) https://doi.org/d8wc48
DOI: 10.1371/journal.pcbi.1000807 · PMID: 20589078 · PMCID: PMC2891586
309. Extracting interactions between proteins from the literature
Deyu Zhou, Yulan He
Journal of Biomedical Informatics (2008-04) https://doi.org/b9kh98
DOI: 10.1016/j.jbi.2007.11.008 · PMID: 18207462
310. Deep learning for extracting protein-protein interactions from
biomedical literature
Yifan Peng, Zhiyong Lu
arXiv (2017-06-05) https://arxiv.org/abs/1706.01556v2
311. DeepPPI: Boosting Prediction of Protein–Protein Interactions with Deep Neural Networks
Xiuquan Du, Shiwei Sun, Changlin Hu, Yu Yao, Yuanting Yan, Yanping Zhang
Journal of Chemical Information and Modeling (2017-05-26) https://doi.org/gbmn43
DOI: 10.1021/acs.jcim.7b00028 · PMID: 28514151
312. Sequence-based prediction of protein protein interaction using a deep-learning algorithm
Tanlin Sun, Bo Zhou, Luhua Lai, Jianfeng Pei
BMC Bioinformatics (2017-05-25) https://doi.org/gcgnzs
DOI: 10.1186/s12859-017-1700-2 · PMID: 28545462 · PMCID: PMC5445391
313. Predicting protein–protein interactions from protein sequences by a stacked sparse autoencoder deep neural network
Yan-Bin Wang, Zhu-Hong You, Xiao Li, Tong-Hai Jiang, Xing Chen, Xi Zhou, Lei Wang
Molecular BioSystems (2017) https://doi.org/gbg8zc
DOI: 10.1039/c7mb00188f · PMID: 28604872
314. Prediction of residue-residue contact matrix for protein-protein interaction with Fisher score features and deep learning
Tianchuan Du, Li Liao, Cathy H. Wu, Bilin Sun
Methods (2016-11) https://doi.org/f9cjkk
DOI: 10.1016/j.ymeth.2016.06.001 · PMID: 27282356
315. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations
Morten Nielsen, Claus Lundegaard, Peder Worning, Sanne Lise Lauemøller, Kasper Lamberth, Søren Buus, Søren Brunak, Ole Lund
Protein Science (2003-05) https://doi.org/bfzjwt
DOI: 10.1110/ps.0239403 · PMID: 12717023 · PMCID: PMC2323871
316. Gapped sequence alignment using artificial neural networks: application to the MHC class I system
Massimo Andreatta, Morten Nielsen
Bioinformatics (2016-02-15) https://doi.org/f8dggc
DOI: 10.1093/bioinformatics/btv639 · PMID: 26515819 · PMCID: PMC6402319
317. NetMHCpan, a method for MHC class I binding prediction beyond humans
Ilka Hoof, Bjoern Peters, John Sidney, Lasse Eggers Pedersen, Alessandro Sette, Ole Lund, Søren Buus, Morten Nielsen
Immunogenetics (2008-11-12) https://doi.org/cn4g24
DOI: 10.1007/s00251-008-0341-z · PMID: 19002680 · PMCID: PMC3319061
318. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets
Morten Nielsen, Massimo Andreatta
Genome Medicine (2016-03-30) https://doi.org/gcgnzt
DOI: 10.1186/s13073-016-0288-x · PMID: 27029192 · PMCID: PMC4812631
319. MHCflurry: open-source class I MHC binding affinity prediction
Timothy O’Donnell, Alex Rubinsteyn, Maria Bonsack, Angelika Riemer, Jeff Hammerbacher
bioRxiv (2017-08-09) https://doi.org/gcpzg6
DOI: 10.1101/174243
320. Predicting Peptide-MHC Binding Affinities with Imputed Training Data
Alex Rubinsteyn, Timothy O’Donnell, Nandita Damaraju, Jeff Hammerbacher
bioRxiv (2016-06-07) https://doi.org/gcgk89
DOI: 10.1101/054775
321. High-order neural networks and kernel methods for peptide-MHC binding prediction
Pavel P. Kuksa, Martin Renqiang Min, Rishabh Dugar, Mark Gerstein
Bioinformatics (2015-07-23) https://doi.org/f7zdkw
DOI: 10.1093/bioinformatics/btv371 · PMID: 26206306
322. Evaluation of machine learning methods to predict peptide binding to MHC Class I proteins
Rohit Bhattacharya, Ashok Sivakumar, Collin Tokheim, Violeta Beleva Guthrie, Valsamo Anagnostou, Victor E. Velculescu, Rachel Karchin
bioRxiv (2017-07-27) https://doi.org/gcpzg5
DOI: 10.1101/154757
323. HLA class I binding prediction via convolutional neural networks
Yeeleng S Vang, Xiaohui Xie
Bioinformatics (2017-09-01) https://doi.org/f94p2v
DOI: 10.1093/bioinformatics/btx264 · PMID: 28444127
324. Network‐based prediction of protein function
Roded Sharan, Igor Ulitsky, Ron Shamir
Molecular Systems Biology (2007-03-13) https://doi.org/cc936j
DOI: 10.1038/msb4100129 · PMID: 17353930 · PMCID: PMC1847944
325. Learning the Structural Vocabulary of a Network
Saket Navlakha
Neural Computation (2017-02) https://doi.org/f9vx8v
DOI: 10.1162/neco_a_00924 · PMID: 28030777
326. deepNF: Deep network fusion for protein function prediction
Vladimir Gligorijević, Meet Barot, Richard Bonneau
bioRxiv (2017-11-22) https://doi.org/gcpzg7
DOI: 10.1101/223339
327. Inductive Representation Learning on Large Graphs
William L. Hamilton, Rex Ying, Jure Leskovec
arXiv (2017-06-07) https://arxiv.org/abs/1706.02216v2
328. Stochastic Training of Graph Convolutional Networks
Jianfei Chen, Jun Zhu
arXiv (2017-10-29) https://arxiv.org/abs/1710.10568v1
329. Deep Learning Automates the Quantitative Analysis of Individual Cells in Live-Cell Imaging Experiments
David A. Van Valen, Takamasa Kudo, Keara M. Lane, Derek N. Macklin, Nicolas T. Quach, Mialy M. DeFelice, Inbal Maayan, Yu Tanouchi, Euan A. Ashley, Markus W. Covert
PLOS Computational Biology (2016-11-04) https://doi.org/f9rfj3
DOI: 10.1371/journal.pcbi.1005177 · PMID: 27814364 · PMCID: PMC5096676
330. U-Net: Convolutional Networks for Biomedical Image Segmentation
Olaf Ronneberger, Philipp Fischer, Thomas Brox
Lecture Notes in Computer Science (2015) https://doi.org/gcgk7j
DOI: 10.1007/978-3-319-24574-4_28
331. Prospective identification of hematopoietic lineage choice by deep learning
Felix Buggenthin, Florian Buettner, Philipp S Hoppe, Max Endele, Manuel Kroiss, Michael Strasser, Michael Schwarzfischer, Dirk Loeffler, Konstantinos D Kokkaliaris, Oliver Hilsenbeck, … Carsten Marr
Nature Methods (2017-02-20) https://doi.org/gcgk8j
DOI: 10.1038/nmeth.4182 · PMID: 28218899 · PMCID: PMC5376497
332. Reconstructing cell cycle and disease progression using deep learning
Philipp Eulenberg, Niklas Köhler, Thomas Blasi, Andrew Filby, Anne E. Carpenter, Paul Rees, Fabian J. Theis, F. Alexander Wolf
bioRxiv (2017-06-05) https://doi.org/gcgk9f
DOI: 10.1101/081364
333. Automating Morphological Profiling with Generic Deep Convolutional Networks
Nick Pawlowski, Juan C Caicedo, Shantanu Singh, Anne E Carpenter, Amos Storkey
bioRxiv (2016-11-02) https://doi.org/gcgk9j
DOI: 10.1101/085118
334. Generative Modeling with Conditional Autoencoders: Building an Integrated Cell
Gregory R. Johnson, Rory M. Donovan-Maiye, Mary M. Maleckar
arXiv (2017-05-02) https://arxiv.org/abs/1705.00092
335. Applications in image-based profiling of perturbations
Juan C Caicedo, Shantanu Singh, Anne E Carpenter
Current Opinion in Biotechnology (2016-06) https://doi.org/f8s4hm
DOI: 10.1016/j.copbio.2016.04.003 · PMID: 27089218
336. Large-scale image-based screening and profiling of cellular phenotypes
Nicola Bougen-Zhukov, Sheng Yang Loh, Hwee Kuan Lee, Lit-Hsin Loo
Cytometry Part A (2017-02) https://doi.org/f9rp9b
DOI: 10.1002/cyto.a.22909 · PMID: 27434125
337. Machine learning and computer vision approaches for phenotypic profiling
Ben T. Grys, Dara S. Lo, Nil Sahin, Oren Z. Kraus, Quaid Morris, Charles Boone, Brenda J. Andrews
The Journal of Cell Biology (2017-01-02) https://doi.org/gcgk8t
DOI: 10.1083/jcb.201610026 · PMID: 27940887 · PMCID: PMC5223612
338. Single-cell genome sequencing: current state of the science
Charles Gawad, Winston Koh, Stephen R. Quake
Nature Reviews Genetics (2016-01-25) https://doi.org/f8ddqt
DOI: 10.1038/nrg.2015.16 · PMID: 26806412
339. Somatic mutation in single human neurons tracks developmental and transcriptional history
Michael A. Lodato, Mollie B. Woodworth, Semin Lee, Gilad D. Evrony, Bhaven K. Mehta, Amir Karger, Soohyun Lee, Thomas W. Chittenden, Alissa M. D’Gama, Xuyu Cai, … Christopher A. Walsh
Science (2015-10-02) https://doi.org/f7sxdv
DOI: 10.1126/science.aab1785 · PMID: 26430121 · PMCID: PMC4664477
340. Single-cell transcriptome sequencing: recent advances and remaining challenges
Serena Liu, Cole Trapnell
F1000Research (2016-02-17) https://doi.org/gcgmcd
DOI: 10.12688/f1000research.7223.1 · PMID: 26949524 · PMCID: PMC4758375
341. Single-Cell and Single-Molecule Analysis of Gene Expression Regulation
Maria Vera, Jeetayu Biswas, Adrien Senecal, Robert H. Singer, Hye Yoon Park
Annual Review of Genetics (2016-11-23) https://doi.org/gcgmb5
DOI: 10.1146/annurev-genet-120215-034854 · PMID: 27893965 · PMCID: PMC5149423
342. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells
Stephen J. Clark, Ricard Argelaguet, Chantriolnt-Andreas Kapourani, Thomas M. Stubbs, Heather J. Lee, Celia Alda-Catalinas, Felix Krueger, Guido Sanguinetti, Gavin Kelsey, John C. Marioni, … Wolf Reik
bioRxiv (2018-01-17) https://doi.org/gcgk93
DOI: 10.1101/138685
343. Denoising Genome-wide Histone ChIP-seq with Convolutional Neural Networks
Pang Wei Koh, Emma Pierson, Anshul Kundaje
bioRxiv (2017-01-27) https://doi.org/gcgk88
DOI: 10.1101/052118
344. Removal of batch effects using distribution-matching residual networks
Uri Shaham, Kelly P Stanton, Jun Zhao, Huamin Li, Khadir Raddassi, Ruth Montgomery, Yuval Kluger
Bioinformatics (2017-08-15) https://doi.org/gbrshc
DOI: 10.1093/bioinformatics/btx196 · PMID: 28419223 · PMCID: PMC5870543
345. Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity
Jellert T. Gaublomme, Nir Yosef, Youjin Lee, Rona S. Gertner, Li V. Yang, Chuan Wu, Pier Paolo Pandolfi, Tak Mak, Rahul Satija, Alex K. Shalek, … Aviv Regev
Cell (2015-12) https://doi.org/f73pfp
DOI: 10.1016/j.cell.2015.11.009 · PMID: 26607794 · PMCID: PMC4671824
346. Sensitive detection of rare disease-associated cell subsets via representation learning
Eirini Arvaniti, Manfred Claassen
bioRxiv (2016-03-31) https://doi.org/gcgk87
DOI: 10.1101/046508
347. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models
Jiarui Ding, Anne Condon, Sohrab P. Shah
bioRxiv (2017-09-01) https://doi.org/gcnzb8
DOI: 10.1101/178624
348. A deep generative model for gene expression profiles from single-cell RNA sequencing
Romain Lopez, Jeffrey Regier, Michael Cole, Michael Jordan, Nir Yosef
arXiv (2018-01-18) https://arxiv.org/abs/1709.02082
349. Using neural networks for reducing the dimensions of single-cell RNA-Seq data
Chieh Lin, Siddhartha Jain, Hannah Kim, Ziv Bar-Joseph
Nucleic Acids Research (2017-09-29) https://doi.org/gcnzb7
DOI: 10.1093/nar/gkx681 · PMID: 28973464 · PMCID: PMC5737331
350. The Human Cell Atlas
Aviv Regev, Sarah A Teichmann, Eric S Lander, Ido Amit, Christophe Benoist, Ewan Birney, Bernd Bodenmiller, Peter Campbell, Piero Carninci, Menna Clatworthy, … Human Cell Atlas Meeting Participants
eLife (2017-12-05) https://doi.org/gcnzcv
DOI: 10.7554/elife.27041 · PMID: 29206104 · PMCID: PMC5762154
351. Reversed graph embedding resolves complex single-cell developmental trajectories
Xiaojie Qiu, Qi Mao, Ying Tang, Li Wang, Raghav Chawla, Hannah Pliner, Cole Trapnell
bioRxiv (2017-02-21) https://doi.org/gcgk9x
DOI: 10.1101/110668
352. Mastering the game of Go with deep neural networks and tree search
David Silver, Aja Huang, Chris J. Maddison, Arthur Guez, Laurent Sifre, George van den Driessche, Julian Schrittwieser, Ioannis Antonoglou, Veda Panneershelvam, Marc Lanctot, … Demis Hassabis
Nature (2016-01-27) https://doi.org/f77tw6
DOI: 10.1038/nature16961 · PMID: 26819042
353. Compositional biases of bacterial genomes and evolutionary implications.
S Karlin, J Mrázek, AM Campbell
Journal of bacteriology (1997) https://doi.org/gcgmbp
DOI: 10.1128/jb.179.12.3899-3913.1997 · PMID: 9190805 · PMCID: PMC179198
354. Accurate phylogenetic classification of variable-length DNA fragments
Alice Carolyn McHardy, Héctor García Martín, Aristotelis Tsirigos, Philip Hugenholtz, Isidore Rigoutsos
Nature Methods (2006-12-10) https://doi.org/fwrtm4
DOI: 10.1038/nmeth976 · PMID: 17179938
355. NBC: the Naive Bayes Classification tool webserver for taxonomic classification of metagenomic reads
G. L. Rosen, E. R. Reichenberger, A. M. Rosenfeld
Bioinformatics (2010-11-08) https://doi.org/dtqrnt
DOI: 10.1093/bioinformatics/btq619 · PMID: 21062764 · PMCID: PMC3008645
356. Informatics for Unveiling Hidden Genome Signatures
T. Abe
Genome Research (2003-04-01) https://doi.org/c475s4
DOI: 10.1101/gr.634603 · PMID: 12671005 · PMCID: PMC430167
357. Metagenomic microbial community profiling using unique clade-specific marker genes
Nicola Segata, Levi Waldron, Annalisa Ballarini, Vagheesh Narasimhan, Olivier Jousson, Curtis Huttenhower
Nature Methods (2012-06-10) https://doi.org/gcgk8f
DOI: 10.1038/nmeth.2066 · PMID: 22688413 · PMCID: PMC3443552
358. WGSQuikr: Fast Whole-Genome Shotgun Metagenomic Classification
David Koslicki, Simon Foucart, Gail Rosen
PLoS ONE (2014-03-13) https://doi.org/gcgmcm
DOI: 10.1371/journal.pone.0091784 · PMID: 24626336 · PMCID: PMC3953531
359. Scalable metagenomic taxonomy classification using a reference genome database
Sasha K. Ames, David A. Hysom, Shea N. Gardner, G. Scott Lloyd, Maya B. Gokhale, Jonathan E. Allen
Bioinformatics (2013-09-15) https://doi.org/f48vw6
DOI: 10.1093/bioinformatics/btt389 · PMID: 23828782 · PMCID: PMC3753567
360. Large-scale machine learning for metagenomics sequence classification
Kévin Vervier, Pierre Mahé, Maud Tournoud, Jean-Baptiste Veyrieras, Jean-Philippe Vert
Bioinformatics (2016-04-01) https://doi.org/f8h92j
DOI: 10.1093/bioinformatics/btv683 · PMID: 26589281 · PMCID: PMC4896366
361. Combining gene prediction methods to improve metagenomic gene annotation
Non G Yok, Gail L Rosen
BMC Bioinformatics (2011-01-13) https://doi.org/dcnc32
DOI: 10.1186/1471-2105-12-20 · PMID: 21232129 · PMCID: PMC3042383
362. Machine learning for metagenomics: methods and tools
Hayssam Soueidan, Macha Nikolski
Metagenomics (2017-01-01) https://doi.org/gcgmct
DOI: 10.1515/metgen-2016-0001
363. Utilizing Machine Learning Approaches to Understand the Interrelationship of Diet, the Human Gastrointestinal Microbiome, and Health
Heather Guetterman, Loretta Auvil, Nate Russell, Michael Welge, Matt Berry, Lisa Gatzke, Colleen Bushell, Hannah Holscher
The FASEB Journal (2016-04-01) https://www.fasebj.org/doi/abs/10.1096/fasebj.30.1_supplement.406.3
364. Supervised classification of human microbiota
Dan Knights, Elizabeth K. Costello, Rob Knight
FEMS Microbiology Reviews (2011-03) https://doi.org/c4bwxs
DOI: 10.1111/j.1574-6976.2010.00251.x · PMID: 21039646
365. A comprehensive evaluation of multicategory classification methods for microbiomic data
Alexander Statnikov, Mikael Henaff, Varun Narendra, Kranti Konganti, Zhiguo Li, Liying Yang, Zhiheng Pei, Martin J Blaser, Constantin F Aliferis, Alexander V Alekseyenko
Microbiome (2013-04-05) https://doi.org/gcgmb6
DOI: 10.1186/2049-2618-1-11 · PMID: 24456583 · PMCID: PMC3960509
366. Machine Learning Meta-analysis of Large Metagenomic Datasets: Tools and Biological Insights
Edoardo Pasolli, Duy Tin Truong, Faizan Malik, Levi Waldron, Nicola Segata
PLOS Computational Biology (2016-07-11) https://doi.org/gcgmch
DOI: 10.1371/journal.pcbi.1004977 · PMID: 27400279 · PMCID: PMC4939962
367. DectICO: an alignment-free supervised metagenomic classification method based on feature extraction and dynamic selection
Xiao Ding, Fudong Cheng, Changchang Cao, Xiao Sun
BMC Bioinformatics (2015-10-07) https://doi.org/f743hh
DOI: 10.1186/s12859-015-0753-3 · PMID: 26446672 · PMCID: PMC4596415
368. Class Prediction and Feature Selection with Linear Optimization for Metagenomic Count Data
Zhenqiu Liu, Dechang Chen, Li Sheng, Amy Y. Liu
PLoS ONE (2013-03-26) https://doi.org/f4w5df
DOI: 10.1371/journal.pone.0053253 · PMID: 23555553 · PMCID: PMC3608598
369. Fizzy: feature subset selection for metagenomics
Gregory Ditzler, J. Calvin Morrison, Yemin Lan, Gail L. Rosen
BMC Bioinformatics (2015-11-04) https://doi.org/gb8w9s
DOI: 10.1186/s12859-015-0793-8 · PMID: 26538306 · PMCID: PMC4634798
370. A Bootstrap Based Neyman-Pearson Test for Identifying Variable Importance
Gregory Ditzler, Robi Polikar, Gail Rosen
IEEE Transactions on Neural Networks and Learning Systems (2015-04) https://doi.org/f66w3q
DOI: 10.1109/tnnls.2014.2320415 · PMID: 25794384
371. Orphelia: predicting genes in metagenomic sequencing reads
Katharina J. Hoff, Thomas Lingner, Peter Meinicke, Maike Tech
Nucleic Acids Research (2009-07-01) https://doi.org/c6mst6
DOI: 10.1093/nar/gkp327 · PMID: 19429689 · PMCID: PMC2703946
372. FragGeneScan: predicting genes in short and error-prone reads
Mina Rho, Haixu Tang, Yuzhen Ye
Nucleic Acids Research (2010-11) https://doi.org/fjg4kc
DOI: 10.1093/nar/gkq747 · PMID: 20805240 · PMCID: PMC2978382
373. Continuous Distributed Representation of Biological Sequences for Deep Proteomics and Genomics
Ehsaneddin Asgari, Mohammad R. K. Mofrad
PLOS ONE (2015-11-10) https://doi.org/gcgmcq
DOI: 10.1371/journal.pone.0141287 · PMID: 26555596 · PMCID: PMC4640716
374. Fast model-based protein homology detection without alignment
S. Hochreiter, M. Heusel, K. Obermayer
Bioinformatics (2007-05-08) https://doi.org/b38n9w
DOI: 10.1093/bioinformatics/btm247 · PMID: 17488755
375. Convolutional LSTM Networks for Subcellular Localization of Proteins
Søren Kaae Sønderby, Casper Kaae Sønderby, Henrik Nielsen, Ole Winther
Lecture Notes in Computer Science (2015) https://doi.org/gcrnp2
DOI: 10.1007/978-3-319-21233-3_6
376. Neural network-based taxonomic clustering for metagenomics
Steven D. Essinger, Robi Polikar, Gail L. Rosen
Institute of Electrical and Electronics Engineers (IEEE) (2010-07) https://doi.org/bnsxts
DOI: 10.1109/ijcnn.2010.5596644
377. Clustering metagenomic sequences with interpolated Markov models
David R Kelley, Steven L Salzberg
BMC Bioinformatics (2010-11-02) https://doi.org/fgjq98
DOI: 10.1186/1471-2105-11-544 · PMID: 21044341 · PMCID: PMC3098094
378. METAGENOMIC TAXONOMIC CLASSIFICATION USING EXTREME LEARNING MACHINES
ZEEHASHAM RASHEED, HUZEFA RANGWALA
Journal of Bioinformatics and Computational Biology (2012-08) https://doi.org/gcgmbt
DOI: 10.1142/s0219720012500151 · PMID: 22849369
379. Globoko ucenje na genomskih in filogenetskih podatkih
Nina Mrzelj
Univerza v Ljubljani, Fakulteta za racunalništvo in informatiko (2016) https://repozitorij.uni-lj.si/IzpisGradiva.php?id=85515
380. Influence of microbiome species in hard-to-heal wounds on disease severity and treatment duration
Dagmar Chudobova, Kristyna Cihalova, Roman Guran, Simona Dostalova, Kristyna Smerkova, Radek Vesely, Jaromir Gumulec, Michal Masarik, Zbynek Heger, Vojtech Adam, Rene Kizek
The Brazilian Journal of Infectious Diseases (2015-11) https://doi.org/gcgk7z
DOI: 10.1016/j.bjid.2015.08.013 · PMID: 26518264
381. Multi-Layer and Recursive Neural Networks for Metagenomic Classification
Gregory Ditzler, Robi Polikar, Gail Rosen
IEEE Transactions on NanoBioscience (2015-09) https://doi.org/gcgmbj
DOI: 10.1109/tnb.2015.2461219 · PMID: 26316190
382. TensorFlow vs. scikit-learn : The Microbiome Challenge
Ali A. Faruqi
Ali A. Faruqi http://alifar76.github.io/sklearn-metrics/
383. Advances in Optimizing Recurrent Networks
Yoshua Bengio, Nicolas Boulanger-Lewandowski, Razvan Pascanu
arXiv (2012-12-04) https://arxiv.org/abs/1212.0901v2
384. DeepNano: Deep recurrent neural networks for base calling in MinION nanopore reads
Vladimír Boža, Broňa Brejová, Tomáš Vinař
PLOS ONE (2017-06-05) https://doi.org/gcrnp4
DOI: 10.1371/journal.pone.0178751 · PMID: 28582401 · PMCID: PMC5459436
385. Sequence to Sequence Learning with Neural Networks
Ilya Sutskever, Oriol Vinyals, Quoc V. Le
arXiv (2014-12-16) https://arxiv.org/abs/1409.3215
386. Creating a universal SNP and small indel variant caller with deep neural networks
Ryan Poplin, Pi-Chuan Chang, David Alexander, Scott Schwartz, Thomas Colthurst, Alexander Ku, Dan Newburger, Jojo Dijamco, Nam Nguyen, Pegah T. Afshar, … Mark A. DePristo
bioRxiv (2018-03-20) https://doi.org/gcgk9m
DOI: 10.1101/092890
387. A framework for variation discovery and genotyping using next-generation DNA sequencing data
Mark A DePristo, Eric Banks, Ryan Poplin, Kiran V Garimella, Jared R Maguire, Christopher Hartl, Anthony A Philippakis, Guillermo del Angel, Manuel A Rivas, Matt Hanna, … Mark J Daly
Nature Genetics (2011-04-10) https://doi.org/d9k453
DOI: 10.1038/ng.806 · PMID: 21478889 · PMCID: PMC3083463
388. Training Genotype Callers with Neural Networks
Remi Torracinta, Fabien Campagne
bioRxiv (2016-12-30) https://doi.org/gcgk9s
DOI: 10.1101/097469
389. Xception: Deep Learning with Depthwise Separable Convolutions
François Chollet
arXiv (2017-04-06) https://arxiv.org/abs/1610.02357
390. Adaptive Somatic Mutations Calls with Deep Learning and Semi-Simulated Data
Remi Torracinta, Laurent Mesnard, Susan Levine, Rita Shaknovich, Maureen Hanson, Susan Levine
bioRxiv (2016-10-04) https://doi.org/gcgk9d
DOI: 10.1101/079087
391. CONTINUATION: Evaluation of adaptive somatic models in a gold standard whole genome somatic dataset
Fabien Campagne
bioRxiv (2016-12-13) https://doi.org/gf5gxz
DOI: 10.1101/093534
392. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing
Tyler S. Alioto, Ivo Buchhalter, Sophia Derdak, Barbara Hutter, Matthew D. Eldridge, Eivind Hovig, Lawrence E. Heisler, Timothy A. Beck, Jared T. Simpson, Laurie Tonon, … Ivo G. Gut
Nature Communications (2015-12-09) https://doi.org/f759g3
DOI: 10.1038/ncomms10001 · PMID: 26647970 · PMCID: PMC4682041
393. Toward an Integration of Deep Learning and Neuroscience
Adam H. Marblestone, Greg Wayne, Konrad P. Kording
Frontiers in Computational Neuroscience (2016-09-14) https://doi.org/gcsgr2
DOI: 10.3389/fncom.2016.00094 · PMID: 27683554 · PMCID: PMC5021692
394. Deep Neural Networks in Computational Neuroscience
Tim C Kietzmann, Patrick McClure, Nikolaus Kriegeskorte
bioRxiv (2018-06-05) https://doi.org/gcsgrx
DOI: 10.1101/133504
395. Neuroscience-Inspired Artificial Intelligence
Demis Hassabis, Dharshan Kumaran, Christopher Summerfield, Matthew Botvinick
Neuron (2017-07) https://doi.org/gbp987
DOI: 10.1016/j.neuron.2017.06.011 · PMID: 28728020
396. Using goal-driven deep learning models to understand sensory cortex
Daniel LK Yamins, James J DiCarlo
Nature Neuroscience (2016-02-23) https://doi.org/gcsgrw
DOI: 10.1038/nn.4244 · PMID: 26906502
397. Inferring single-trial neural population dynamics using sequential auto-encoders
Chethan Pandarinath, Daniel J. O’Shea, Jasmine Collins, Rafal Jozefowicz, Sergey D. Stavisky, Jonathan C. Kao, Eric M. Trautmann, Matthew T. Kaufman, Stephen I. Ryu, Leigh R. Hochberg, … David Sussillo
bioRxiv (2017-06-20) https://doi.org/gcsgrz
DOI: 10.1101/152884
398. Machines that learn to segment images: a crucial technology for connectomics
Viren Jain, H Sebastian Seung, Srinivas C Turaga
Current Opinion in Neurobiology (2010-10) https://doi.org/b23662
DOI: 10.1016/j.conb.2010.07.004 · PMID: 20801638 · PMCID: PMC2975605
399. Model-based Bayesian inference of neural activity and connectivity from all-optical interrogation of a neural circuit
Laurence Aitchison, Lloyd Russell, Adam M Packer, Jinyao Yan, Philippe Castonguay, Michael Hausser, Srinivas C Turaga
Advances in Neural Information Processing Systems 30 (2017) http://papers.nips.cc/paper/6940-model-based-bayesian-inference-of-neural-activity-and-connectivity-from-all-optical-interrogation-of-a-neural-circuit.pdf
400. The Path to Personalized Medicine
Margaret A. Hamburg, Francis S. Collins
New England Journal of Medicine (2010-07-22) https://doi.org/bp78cs
DOI: 10.1056/nejmp1006304 · PMID: 20551152
401. Biomedical Informatics for Computer-Aided Decision Support Systems: A Survey
Ashwin Belle, Mark A. Kon, Kayvan Najarian
The Scientific World Journal (2013) https://doi.org/gb7x73
DOI: 10.1155/2013/769639 · PMID: 23431259 · PMCID: PMC3575619
402. Advantages and disadvantages of using artificial neural networks versus logistic regression for predicting medical outcomes
Jack V. Tu
Journal of Clinical Epidemiology (1996-11) https://doi.org/fg794g
DOI: 10.1016/s0895-4356(96)00002-9
403. Use of an Artificial Neural Network for the Diagnosis of Myocardial Infarction
William G. Baxt
Annals of Internal Medicine (1991-12-01) https://doi.org/gcgmc5
DOI: 10.7326/0003-4819-115-11-843 · PMID: 1952470
404. Clinical Prediction Rules
John H. Wasson, Harold C. Sox, Raymond K. Neff, Lee Goldman
New England Journal of Medicine (1985-09-26) https://doi.org/d7mchc
DOI: 10.1056/nejm198509263131306 · PMID: 3897864
405. The use of artificial neural networks in decision support in cancer: A systematic review
Paulo J. Lisboa, Azzam F. G. Taktak
Neural Networks (2006-05) https://doi.org/cnvgmv
DOI: 10.1016/j.neunet.2005.10.007 · PMID: 16483741
406. Estimating causal effects of treatments in randomized and nonrandomized studies.
Donald B. Rubin
Journal of Educational Psychology (1974) https://doi.org/cqq7sc
DOI: 10.1037/h0037350
407. Learning Representations for Counterfactual Inference
Fredrik D. Johansson, Uri Shalit, David Sontag
arXiv (2018-06-07) https://arxiv.org/abs/1605.03661
408. Causal Phenotype Discovery via Deep Networks
David C Kale, Zhengping Che, Mohammad Taha Bahadori, Wenzhe Li, Yan Liu, Randall Wetzel
AMIA … Annual Symposium proceedings. AMIA Symposium (2015-11-05) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4765623/
PMID: 26958203 · PMCID: PMC4765623
409. Modeling Missing Data in Clinical Time Series with RNNs
Zachary C. Lipton, David C. Kale, Randall Wetzel
arXiv (2016-11-14) https://arxiv.org/abs/1606.04130
410. Recurrent Neural Networks for Multivariate Time Series with Missing Values
Zhengping Che, Sanjay Purushotham, Kyunghyun Cho, David Sontag, Yan Liu
arXiv (2016-11-08) https://arxiv.org/abs/1606.01865
411. Predicting Complications in Critical Care Using Heterogeneous Clinical Data
Vijay Huddar, Bapu Koundinya Desiraju, Vaibhav Rajan, Sakyajit Bhattacharya, Shourya Roy, Chandan K. Reddy
IEEE Access (2016) https://doi.org/gcgk94
DOI: 10.1109/access.2016.2618775
412. Phenotyping of Clinical Time Series with LSTM Recurrent Neural Networks
Zachary C. Lipton, David C. Kale, Randall C. Wetzel
arXiv (2017-03-23) https://arxiv.org/abs/1510.07641
413. Optimal medication dosing from suboptimal clinical examples: A deep reinforcement learning approach
Shamim Nemati, Mohammad M. Ghassemi, Gari D. Clifford
Institute of Electrical and Electronics Engineers (IEEE) (2016-08) https://doi.org/gcgk98
DOI: 10.1109/embc.2016.7591355 · PMID: 28268938
414. From vital signs to clinical outcomes for patients with sepsis: a machine learning basis for a clinical decision support system
Eren Gultepe, Jeffrey P Green, Hien Nguyen, Jason Adams, Timothy Albertson, Ilias Tagkopoulos
Journal of the American Medical Informatics Association (2014-03-01) https://doi.org/f5trc2
DOI: 10.1136/amiajnl-2013-001815 · PMID: 23959843 · PMCID: PMC3932455
415. Imaging-based enrichment criteria using deep learning algorithms for efficient clinical trials in mild cognitive impairment
Vamsi K. Ithapu, Vikas Singh, Ozioma C. Okonkwo, Richard J. Chappell, N. Maritza Dowling, Sterling C. Johnson, Alzheimer’s Disease Neuroimaging Initiative
Alzheimer’s & Dementia (2015-12) https://doi.org/f73fvf
DOI: 10.1016/j.jalz.2015.01.010 · PMID: 26093156 · PMCID: PMC4684492
416. Integrated deep learned transcriptomic and structure-based predictor of clinical trials outcomes
Artem V. Artemov, Evgeny Putin, Quentin Vanhaelen, Alexander Aliper, Ivan V. Ozerov, Alex Zhavoronkov
bioRxiv (2016-12-29) https://doi.org/gcgk9p
DOI: 10.1101/095653
417. Innovation in the pharmaceutical industry: New estimates of R&D costs
Joseph A. DiMasi, Henry G. Grabowski, Ronald W. Hansen
Journal of Health Economics (2016-05) https://doi.org/f3mn5k
DOI: 10.1016/j.jhealeco.2016.01.012 · PMID: 26928437
418. An analysis of the attrition of drug candidates from four major pharmaceutical companies
Michael J. Waring, John Arrowsmith, Andrew R. Leach, Paul D. Leeson, Sam Mandrell, Robert M. Owen, Garry Pairaudeau, William D. Pennie, Stephen D. Pickett, Jibo Wang, … Alex Weir
Nature Reviews Drug Discovery (2015-06-19) https://doi.org/f7hqmh
DOI: 10.1038/nrd4609 · PMID: 26091267
419. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease
J. Lamb
Science (2006-09-29) https://doi.org/c92ptt
DOI: 10.1126/science.1132939 · PMID: 17008526
420. A survey of current trends in computational drug repositioning
Jiao Li, Si Zheng, Bin Chen, Atul J. Butte, S. Joshua Swamidass, Zhiyong Lu
Briefings in Bioinformatics (2016-01) https://doi.org/f78wph
DOI: 10.1093/bib/bbv020 · PMID: 25832646 · PMCID: PMC4719067
421. A review of connectivity map and computational approaches in pharmacogenomics
Aliyu Musa, Laleh Soltan Ghoraie, Shu-Dong Zhang, Galina Galzko, Olli Yli-Harja, Matthias Dehmer, Benjamin Haibe-Kains, Frank Emmert-Streib
Briefings in Bioinformatics (2017-01-09) https://doi.org/gcgk8w
DOI: 10.1093/bib/bbw112 · PMID: 28069634 · PMCID: PMC5952941
422. A review of validation strategies for computational drug repositioning
Adam S. Brown, Chirag J. Patel
Briefings in Bioinformatics (2016-11-22) https://academic.oup.com/bib/article/doi/10.1093/bib/bbw110/2562646/A-review-of-validation-strategies-for
DOI: 10.1093/bib/bbw110
423. Drug repositioning: a machine-learning approach through data integration
Francesco Napolitano, Yan Zhao, Vânia M Moreira, Roberto Tagliaferri, Juha Kere, Mauro D’Amato, Dario Greco
Journal of Cheminformatics (2013-06-22) https://doi.org/f243g5
DOI: 10.1186/1758-2946-5-30 · PMID: 23800010 · PMCID: PMC3704944
424. Drug–Disease Association and Drug-Repositioning Predictions in Complex Diseases Using Causal Inference–Probabilistic Matrix Factorization
Jihong Yang, Zheng Li, Xiaohui Fan, Yiyu Cheng
Journal of Chemical Information and Modeling (2014-08-22) https://doi.org/f6hpb4
DOI: 10.1021/ci500340n · PMID: 25116798
425. Drug repositioning for non-small cell lung cancer by using machine learning algorithms and topological graph theory
Chien-Hung Huang, Peter Mu-Hsin Chang, Chia-Wei Hsu, Chi-Ying F. Huang, Ka-Lok Ng
BMC Bioinformatics (2016-01-11) https://doi.org/gcgmb7
DOI: 10.1186/s12859-015-0845-0 · PMID: 26817825 · PMCID: PMC4895785
426. Machine Learning Prediction of Cancer Cell Sensitivity to Drugs Based on Genomic and Chemical Properties
Michael P. Menden, Francesco Iorio, Mathew Garnett, Ultan McDermott, Cyril H. Benes, Pedro J. Ballester, Julio Saez-Rodriguez
PLoS ONE (2013-04-30) https://doi.org/f4vfwd
DOI: 10.1371/journal.pone.0061318 · PMID: 23646105 · PMCID: PMC3640019
427. Large-scale integration of small-molecule induced genome-wide transcriptional responses, Kinome-wide binding affinities and cell-growth inhibition profiles reveal global trends characterizing systems-level drug action
Dušica Vidovic, Amar Koleti, Stephan C Schürer
Frontiers in Genetics https://doi.org/10.3389/fgene.2014.00342
DOI: 10.3389/fgene.2014.00342
428. Computational Discovery of Putative Leads for Drug Repositioning through Drug-Target Interaction Prediction
Edgar D. Coelho, Joel P. Arrais, José Luís Oliveira
PLOS Computational Biology (2016-11-28) https://doi.org/gcgmcj
DOI: 10.1371/journal.pcbi.1005219 · PMID: 27893735 · PMCID: PMC5125559
429. Large-Scale Off-Target Identification Using Fast and Accurate Dual Regularized One-Class Collaborative Filtering and Its Application to Drug Repurposing
Hansaim Lim, Aleksandar Poleksic, Yuan Yao, Hanghang Tong, Di He, Luke Zhuang, Patrick Meng, Lei Xie
PLOS Computational Biology (2016-10-07) https://doi.org/f9ktgd
DOI: 10.1371/journal.pcbi.1005135 · PMID: 27716836 · PMCID: PMC5055357
430. Pairwise input neural network for target-ligand interaction prediction
Caihua Wang, Juan Liu, Fei Luo, Yafang Tan, Zixin Deng, Qian-Nan Hu
Institute of Electrical and Electronics Engineers (IEEE) (2014-11) https://doi.org/gcgk95
DOI: 10.1109/bibm.2014.6999129
431. L1000CDS2: LINCS L1000 characteristic direction signatures search engine
Qiaonan Duan, St Patrick Reid, Neil R Clark, Zichen Wang, Nicolas F Fernandez, Andrew D Rouillard, Ben Readhead, Sarah R Tritsch, Rachel Hodos, Marc Hafner, … Avi Ma’ayan
npj Systems Biology and Applications (2016-08-04) https://doi.org/gcgk8k
DOI: 10.1038/npjsba.2016.15 · PMID: 28413689 · PMCID: PMC5389891
432. Hit and lead generation: beyond high-throughput screening
Konrad H. Bleicher, Hans-Joachim Böhm, Klaus Müller, Alexander I. Alanine
Nature Reviews Drug Discovery (2003-05) https://doi.org/c8crxp
DOI: 10.1038/nrd1086 · PMID: 12750740
433. Hit discovery and hit-to-lead approaches
György M. Keserű, Gergely M. Makara
Drug Discovery Today (2006-08) https://doi.org/ffz3hv
DOI: 10.1016/j.drudis.2006.06.016 · PMID: 16846802
434. Influence Relevance Voting: An Accurate And Interpretable Virtual High Throughput Screening Method
S. Joshua Swamidass, Chloé-Agathe Azencott, Ting-Wan Lin, Hugo Gramajo, Shiou-Chuan Tsai, Pierre Baldi
Journal of Chemical Information and Modeling (2009-03-26) https://doi.org/cw5jfr
DOI: 10.1021/ci8004379 · PMID: 19391629 · PMCID: PMC2750043
435. Modeling Industrial ADMET Data with Multitask Networks
Steven Kearnes, Brian Goldman, Vijay Pande
arXiv (2017-01-16) https://arxiv.org/abs/1606.08793
436. XenoSite: Accurately Predicting CYP-Mediated Sites of Metabolism with Neural Networks
Jed Zaretzki, Matthew Matlock, S. Joshua Swamidass
Journal of Chemical Information and Modeling (2013-11-23) https://doi.org/f5nfz3
DOI: 10.1021/ci400518g · PMID: 24224933
437. Multi-task Neural Networks for QSAR Predictions
George E. Dahl, Navdeep Jaitly, Ruslan Salakhutdinov
arXiv (2014-06-06) https://arxiv.org/abs/1406.1231
438. Deep Neural Nets as a Method for Quantitative Structure–Activity Relationships
Junshui Ma, Robert P. Sheridan, Andy Liaw, George E. Dahl, Vladimir Svetnik
Journal of Chemical Information and Modeling (2015-02-17) https://doi.org/f6358c
DOI: 10.1021/ci500747n · PMID: 25635324
439. Did Kaggle Predict Drug Candidate Activities? Or Not?
Derek Lowe 11 December, 2012
In the Pipeline (2012-12-11) https://blogs.sciencemag.org/pipeline/archives/2012/12/11/did_kaggle_predict_drug_candidate_activities_or_not
440. Deep learning as an opportunity in virtual screening
Thomas Unterthiner, Andreas Mayr, Günter Klambauer, Marvin Steijaert, Jörg K. Wegner, Hugo Ceulemans, Sepp Hochreiter
Neural Information Processing Systems 2014: Deep Learning and Representation Learning Workshop (2014) http://www.dlworkshop.org/23.pdf?attredirects=0
441. Massively Multitask Networks for Drug Discovery
Bharath Ramsundar, Steven Kearnes, Patrick Riley, Dale Webster, David Konerding, Vijay Pande
arXiv (2015-02-10) https://arxiv.org/abs/1502.02072
442. DeepTox: Toxicity Prediction using Deep Learning
Andreas Mayr, Günter Klambauer, Thomas Unterthiner, Sepp Hochreiter
Frontiers in Environmental Science (2016-02-02) https://doi.org/gcgmc3
DOI: 10.3389/fenvs.2015.00080
443. Computational Modeling of β-Secretase 1 (BACE-1) Inhibitors Using Ligand Based Approaches
Govindan Subramanian, Bharath Ramsundar, Vijay Pande, Rajiah Aldrin Denny
Journal of Chemical Information and Modeling (2016-10-10) https://doi.org/f88ngh
DOI: 10.1021/acs.jcim.6b00290 · PMID: 27689393
444. The enumeration of chemical space
Jean-Louis Reymond, Lars Ruddigkeit, Lorenz Blum, Ruud van Deursen
Wiley Interdisciplinary Reviews: Computational Molecular Science (2012-09) https://doi.org/gcgk7f
DOI: 10.1002/wcms.1104
445. Accurate and efficient target prediction using a potency-sensitive influence-relevance voter
Alessandro Lusci, David Fooshee, Michael Browning, Joshua Swamidass, Pierre Baldi
Journal of Cheminformatics (2015-12-29) https://doi.org/f76hzq
DOI: 10.1186/s13321-015-0110-6 · PMID: 26719774 · PMCID: PMC4696267
446. Molecular Descriptors for Chemoinformatics
Roberto Todeschini, Viviana Consonni
Methods and Principles in Medicinal Chemistry (2009-07-15) https://doi.org/c9xk4r
DOI: 10.1002/9783527628766
447. Extended-Connectivity Fingerprints
David Rogers, Mathew Hahn
Journal of Chemical Information and Modeling (2010-04-28) https://doi.org/fp3ctj
DOI: 10.1021/ci100050t · PMID: 20426451
448. Automatic chemical design using a data-driven continuous representation
of molecules
Rafael Gómez-Bombarelli, David Duvenaud, José Miguel Hernández-Lobato, Jorge Aguilera-Iparraguirre, Timothy D. Hirzel, Ryan P. Adams, Alán Aspuru-Guzik
arXiv (2016-10-07) https://arxiv.org/abs/1610.02415v1
449. Chemception: A Deep Neural Network with Minimal Chemistry Knowledge Matches the Performance of Expert-developed QSAR/QSPR Models
Garrett B. Goh, Charles Siegel, Abhinav Vishnu, Nathan O. Hodas, Nathan Baker
arXiv (2018-08-15) https://arxiv.org/abs/1706.06689
450. Convolutional Networks on Graphs for Learning Molecular Fingerprints
David K Duvenaud, Dougal Maclaurin, Jorge Iparraguirre, Rafael Bombarell, Timothy Hirzel, Alan Aspuru-Guzik, Ryan P Adams
Advances in Neural Information Processing Systems 28 (2015) http://papers.nips.cc/paper/5954-convolutional-networks-on-graphs-for-learning-molecular-fingerprints.pdf
451. Deep Architectures and Deep Learning in Chemoinformatics: The Prediction of Aqueous Solubility for Drug-Like Molecules
Alessandro Lusci, Gianluca Pollastri, Pierre Baldi
Journal of Chemical Information and Modeling (2013-07-02) https://doi.org/f48nb7
DOI: 10.1021/ci400187y · PMID: 23795551 · PMCID: PMC3739985
452. Molecular graph convolutions: moving beyond fingerprints
Steven Kearnes, Kevin McCloskey, Marc Berndl, Vijay Pande, Patrick Riley
Journal of Computer-Aided Molecular Design (2016-08-24) https://doi.org/f86bbs
DOI: 10.1007/s10822-016-9938-8 · PMID: 27558503 · PMCID: PMC5028207
453. Low Data Drug Discovery with One-Shot Learning
Han Altae-Tran, Bharath Ramsundar, Aneesh S. Pappu, Vijay Pande
ACS Central Science (2017-04-03) https://doi.org/f95dnd
DOI: 10.1021/acscentsci.6b00367 · PMID: 28470045 · PMCID: PMC5408335
454. Convolutional Embedding of Attributed Molecular Graphs for Physical Property Prediction
Connor W. Coley, Regina Barzilay, William H. Green, Tommi S. Jaakkola, Klavs F. Jensen
Journal of Chemical Information and Modeling (2017-07-25) https://doi.org/gbpqrq
DOI: 10.1021/acs.jcim.6b00601 · PMID: 28696688
455. Learning a Local-Variable Model of Aromatic and Conjugated Systems
Matthew K. Matlock, Na Le Dang, S. Joshua Swamidass
ACS Central Science (2018-01-03) https://doi.org/gcsgr4
DOI: 10.1021/acscentsci.7b00405 · PMID: 29392176 · PMCID: PMC5785769
456. Covariant Compositional Networks For Learning Graphs
Risi Kondor, Hy Truong Son, Horace Pan, Brandon Anderson, Shubhendu Trivedi
arXiv (2018-01-09) https://arxiv.org/abs/1801.02144
457. MoleculeNet: a benchmark for molecular machine learning
Zhenqin Wu, Bharath Ramsundar, Evan N. Feinberg, Joseph Gomes, Caleb Geniesse, Aneesh S. Pappu, Karl Leswing, Vijay Pande
Chemical Science (2018) https://doi.org/gcsgr5
DOI: 10.1039/c7sc02664a · PMID: 29629118 · PMCID: PMC5868307
458. What do we know and when do we know it?
Anthony Nicholls
Journal of Computer-Aided Molecular Design (2008-02-06) https://doi.org/cfp6g6
DOI: 10.1007/s10822-008-9170-2 · PMID: 18253702 · PMCID: PMC2270923
459. deepchem/deepchem
GitHub
(2017) https://github.com/deepchem/deepchem
460. Mol2vec: Unsupervised Machine Learning Approach with Chemical Intuition
Sabrina Jaeger, Simone Fulle, Samo Turk
Journal of Chemical Information and Modeling (2018-01-10) https://doi.org/gcsgwx
DOI: 10.1021/acs.jcim.7b00616 · PMID: 29268609
461. Structure-Based Virtual Screening for Drug Discovery: a Problem-Centric Review
Tiejun Cheng, Qingliang Li, Zhigang Zhou, Yanli Wang, Stephen H. Bryant
The AAPS Journal (2012-01-27) https://doi.org/fxjg96
DOI: 10.1208/s12248-012-9322-0 · PMID: 22281989 · PMCID: PMC3282008
462. Atomic Convolutional Networks for Predicting Protein-Ligand Binding Affinity
Joseph Gomes, Bharath Ramsundar, Evan N. Feinberg, Vijay S. Pande
arXiv (2017-03-31) https://arxiv.org/abs/1703.10603
463. TopologyNet: Topology based deep convolutional and multi-task neural networks for biomolecular property predictions
Zixuan Cang, Guo-Wei Wei
PLOS Computational Biology (2017-07-27) https://doi.org/gbp9ps
DOI: 10.1371/journal.pcbi.1005690 · PMID: 28749969 · PMCID: PMC5549771
464. The PDBbind Database: Methodologies and Updates
Renxiao Wang, Xueliang Fang, Yipin Lu, Chao-Yie Yang, Shaomeng Wang
Journal of Medicinal Chemistry (2005-06) https://doi.org/djbvfc
DOI: 10.1021/jm048957q · PMID: 15943484
465. Boosting Docking-Based Virtual Screening with Deep Learning
Janaina Cruz Pereira, Ernesto Raúl Caffarena, Cicero Nogueira dos Santos
Journal of Chemical Information and Modeling (2016-11-29) https://doi.org/f9jhpn
DOI: 10.1021/acs.jcim.6b00355 · PMID: 28024405
466. Protein-Ligand Scoring with Convolutional Neural Networks
Matthew Ragoza, Joshua Hochuli, Elisa Idrobo, Jocelyn Sunseri, David Ryan Koes
arXiv (2016-12-09) https://arxiv.org/abs/1612.02751
467. Enabling future drug discovery by de novo design
Markus Hartenfeller, Gisbert Schneider
WIREs Computational Molecular Science (2011-04-25) https://doi.org/bv7hkf
DOI: 10.1002/wcms.49
468. De Novo Design at the Edge of Chaos
Petra Schneider, Gisbert Schneider
Journal of Medicinal Chemistry (2016-02-16) https://doi.org/gcgk76
DOI: 10.1021/acs.jmedchem.5b01849 · PMID: 26881908
469. Hunting for Organic Molecules with Artificial Intelligence: Molecules Optimized for Desired Excitation Energies
Masato Sumita, Xiufeng Yang, Shinsuke Ishihara, Ryo Tamura, Koji Tsuda
ACS Central Science (2018-08-20) https://doi.org/gfcpxs
DOI: 10.1021/acscentsci.8b00213 · PMID: 30276245 · PMCID: PMC6161049
470. Deep learning for molecular design—a review of the state of the art
Daniel C. Elton, Zois Boukouvalas, Mark D. Fuge, Peter W. Chung
Molecular Systems Design & Engineering (2019) https://doi.org/gf7hqz
DOI: 10.1039/c9me00039a
471. Applications of machine learning in drug discovery and development
Jessica Vamathevan, Dominic Clark, Paul Czodrowski, Ian Dunham, Edgardo Ferran, George Lee, Bin Li, Anant Madabhushi, Parantu Shah, Michaela Spitzer, Shanrong Zhao
Nature Reviews Drug Discovery (2019-04-11) https://doi.org/gf2w7n
DOI: 10.1038/s41573-019-0024-5 · PMID: 30976107 · PMCID: PMC6552674
472. Generating Sequences With Recurrent Neural Networks
Alex Graves
arXiv (2014-06-06) https://arxiv.org/abs/1308.0850
473. Generating Focussed Molecule Libraries for Drug Discovery with Recurrent Neural Networks
Marwin H. S. Segler, Thierry Kogej, Christian Tyrchan, Mark P. Waller
arXiv (2017-01-06) https://arxiv.org/abs/1701.01329
474. Grammar Variational Autoencoder
Matt J. Kusner, Brooks Paige, José Miguel Hernández-Lobato
arXiv (2017-03-07) https://arxiv.org/abs/1703.01925
475. ChEMBL: a large-scale bioactivity database for drug discovery
A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani, J. P. Overington
Nucleic Acids Research (2011-09-23) https://doi.org/bs9shd
DOI: 10.1093/nar/gkr777 · PMID: 21948594 · PMCID: PMC3245175
476. Molecular De Novo Design through Deep Reinforcement Learning
Marcus Olivecrona, Thomas Blaschke, Ola Engkvist, Hongming Chen
arXiv (2017-08-30) https://arxiv.org/abs/1704.07555
477. Sequence Tutor: Conservative Fine-Tuning of Sequence Generation Models with KL-control
Natasha Jaques, Shixiang Gu, Dzmitry Bahdanau, José Miguel Hernández-Lobato, Richard E. Turner, Douglas Eck
arXiv (2017-10-18) https://arxiv.org/abs/1611.02796
478. Optimization of Molecules via Deep Reinforcement Learning
Zhenpeng Zhou, Steven Kearnes, Li Li, Richard N. Zare, Patrick Riley
Scientific Reports (2019-07-24) https://doi.org/ggfqc8
DOI: 10.1038/s41598-019-47148-x · PMID: 31341196 · PMCID: PMC6656766
479. Junction Tree Variational Autoencoder for Molecular Graph Generation
Wengong Jin, Regina Barzilay, Tommi Jaakkola
arXiv (2019-04-01) https://arxiv.org/abs/1802.04364
480. Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models
Gabriel Lima Guimaraes, Benjamin Sanchez-Lengeling, Carlos Outeiral, Pedro Luis Cunha Farias, Alán Aspuru-Guzik
arXiv (2018-02-08) https://arxiv.org/abs/1705.10843
481. Graph Convolutional Policy Network for Goal-Directed Molecular Graph Generation
Jiaxuan You, Bowen Liu, Rex Ying, Vijay Pande, Jure Leskovec
arXiv (2019-02-26) https://arxiv.org/abs/1806.02473
482. Deep learning enables rapid identification of potent DDR1 kinase inhibitors
Alex Zhavoronkov, Yan A. Ivanenkov, Alex Aliper, Mark S. Veselov, Vladimir A. Aladinskiy, Anastasiya V. Aladinskaya, Victor A. Terentiev, Daniil A. Polykovskiy, Maksim D. Kuznetsov, Arip Asadulaev, … Alán Aspuru-Guzik
Nature Biotechnology (2019-09-02) https://doi.org/gf7gc9
DOI: 10.1038/s41587-019-0224-x · PMID: 31477924
483. Assessing the impact of generative AI on medicinal chemistry
W. Patrick Walters, Mark Murcko
Nature Biotechnology (2020-01-30) https://doi.org/ggkmqn
DOI: 10.1038/s41587-020-0418-2 · PMID: 32001834
484. GuacaMol: Benchmarking Models for de Novo Molecular Design
Nathan Brown, Marco Fiscato, Marwin H. S. Segler, Alain C. Vaucher
Journal of Chemical Information and Modeling (2019-03-19) https://doi.org/ggpn3x
DOI: 10.1021/acs.jcim.8b00839 · PMID: 30887799
485. The Synthesizability of Molecules Proposed by Generative Models
Wenhao Gao, Connor W. Coley
Journal of Chemical Information and Modeling (2020-04-06) https://doi.org/gg7gx7
DOI: 10.1021/acs.jcim.0c00174 · PMID: 32250616
486. The Advent of Generative Chemistry
Quentin Vanhaelen, Yen-Chu Lin, Alex Zhavoronkov
ACS Medicinal Chemistry Letters (2020-07-14) https://doi.org/gg7hmb
DOI: 10.1021/acsmedchemlett.0c00088 · PMID: 32832015 · PMCID: PMC7429972
487. A graph-based genetic algorithm and generative model/Monte Carlo tree search for the exploration of chemical space
Jan H. Jensen
Chemical Science (2019) https://doi.org/ggfqc9
DOI: 10.1039/c8sc05372c · PMID: 30996948 · PMCID: PMC6438151
488. Population-based De Novo Molecule Generation, Using Grammatical Evolution
Naruki Yoshikawa, Kei Terayama, Masato Sumita, Teruki Homma, Kenta Oono, Koji Tsuda
Chemistry Letters (2018-11-05) https://doi.org/ggfqdb
DOI: 10.1246/cl.180665
489. Understanding deep learning requires rethinking generalization
Chiyuan Zhang, Samy Bengio, Moritz Hardt, Benjamin Recht, Oriol Vinyals
arXiv (2016-11-10) https://arxiv.org/abs/1611.03530v2
490. Why does deep and cheap learning work so well?
Henry W. Lin, Max Tegmark, David Rolnick
arXiv (2016-08-29) https://arxiv.org/abs/1608.08225v3
491. The relationship between Precision-Recall and ROC curves
Jesse Davis, Mark Goadrich
Association for Computing Machinery (ACM) (2006) https://doi.org/fc8wzr
DOI: 10.1145/1143844.1143874
492. An open investigation of the reproducibility of cancer biology research
Timothy M Errington, Elizabeth Iorns, William Gunn, Fraser Elisabeth Tan, Joelle Lomax, Brian A Nosek
eLife (2014-12-10) https://doi.org/gcnzrq
DOI: 10.7554/elife.04333 · PMID: 25490932 · PMCID: PMC4270077
493. Adversarial Examples, Uncertainty, and Transfer Testing Robustness in Gaussian Process Hybrid Deep Networks
John Bradshaw, Alexander G. de G. Matthews, Zoubin Ghahramani
arXiv (2017-07-11) https://arxiv.org/abs/1707.02476
494. What Uncertainties Do We Need in Bayesian Deep Learning for Computer Vision?
Alex Kendall, Yarin Gal
arXiv (2017-10-06) https://arxiv.org/abs/1703.04977
495. Multi-Task Learning Using Uncertainty to Weigh Losses for Scene Geometry and Semantics
Alex Kendall, Yarin Gal, Roberto Cipolla
arXiv (2018-04-25) https://arxiv.org/abs/1705.07115
496. On Calibration of Modern Neural Networks
Chuan Guo, Geoff Pleiss, Yu Sun, Kilian Q. Weinberger
arXiv (2017-08-04) https://arxiv.org/abs/1706.04599
497. Probabilistic Outputs for Support Vector Machines and Comparisons to Regularized Likelihood Methods
John C. Platt
Advances in Large Margin Classifiers (1999)
498. Confidence interval prediction for neural network models
G. Chryssolouris, M. Lee, A. Ramsey
IEEE Transactions on Neural Networks https://doi.org/cfc8m8
DOI: 10.1109/72.478409 · PMID: 18255575
499. A Baseline for Detecting Misclassified and Out-of-Distribution Examples in Neural Networks
Dan Hendrycks, Kevin Gimpel
arXiv (2018-10-04) https://arxiv.org/abs/1610.02136
500. Enhancing The Reliability of Out-of-distribution Image Detection in Neural Networks
Shiyu Liang, Yixuan Li, R. Srikant
arXiv (2018-02-27) https://arxiv.org/abs/1706.02690
501. Concrete Problems in AI Safety
Dario Amodei, Chris Olah, Jacob Steinhardt, Paul Christiano, John Schulman, Dan Mané
arXiv (2016-07-26) https://arxiv.org/abs/1606.06565
502. Adversarial Examples Are Not Easily Detected: Bypassing Ten Detection Methods
Nicholas Carlini, David Wagner
arXiv (2017-11-02) https://arxiv.org/abs/1705.07263
503. Dropout as a Bayesian Approximation: Representing Model Uncertainty in Deep Learning
Yarin Gal, Zoubin Ghahramani
arXiv (2016-10-05) https://arxiv.org/abs/1506.02142
504. Leveraging uncertainty information from deep neural networks for disease detection
Christian Leibig, Vaneeda Allken, Murat Seçkin Ayhan, Philipp Berens, Siegfried Wahl
Scientific Reports (2017-12-19) https://doi.org/gcqgdc
DOI: 10.1038/s41598-017-17876-z · PMID: 29259224 · PMCID: PMC5736701
505. Robustly representing uncertainty in deep neural networks through sampling
Patrick McClure, Nikolaus Kriegeskorte
arXiv (2019-09-19) https://arxiv.org/abs/1611.01639
506. Bayesian Hypernetworks
David Krueger, Chin-Wei Huang, Riashat Islam, Ryan Turner, Alexandre Lacoste, Aaron Courville
arXiv (2018-04-26) https://arxiv.org/abs/1710.04759
507. Simple and Scalable Predictive Uncertainty Estimation using Deep Ensembles
Balaji Lakshminarayanan, Alexander Pritzel, Charles Blundell
arXiv (2017-11-07) https://arxiv.org/abs/1612.01474
508. Uncertainty in deep learning
Yarin Gal
PhD thesis, University of Cambridge (2016) http://www.cs.ox.ac.uk/people/yarin.gal/website/thesis/thesis.pdf
509. Intelligible Models for HealthCare
Rich Caruana, Yin Lou, Johannes Gehrke, Paul Koch, Marc Sturm, Noemie Elhadad
Association for Computing Machinery (ACM) (2015) https://doi.org/gftgxk
DOI: 10.1145/2783258.2788613
510. Deep Neural Networks are Easily Fooled: High Confidence Predictions for
Unrecognizable Images
Anh Nguyen, Jason Yosinski, Jeff Clune
arXiv (2014-12-05) https://arxiv.org/abs/1412.1897v4
511. “Why Should I Trust You?”: Explaining the Predictions of Any Classifier
Marco Tulio Ribeiro, Sameer Singh, Carlos Guestrin
arXiv (2016-08-10) https://arxiv.org/abs/1602.04938
512. Visualizing and Understanding Convolutional Networks
Matthew D Zeiler, Rob Fergus
arXiv (2013-12-02) https://arxiv.org/abs/1311.2901
513. Visualizing Deep Neural Network Decisions: Prediction Difference Analysis
Luisa M Zintgraf, Taco S Cohen, Tameem Adel, Max Welling
arXiv (2017-02-16) https://arxiv.org/abs/1702.04595
514. Interpretable Explanations of Black Boxes by Meaningful Perturbation
Ruth C. Fong, Andrea Vedaldi
Institute of Electrical and Electronics Engineers (IEEE) (2017-10) https://doi.org/gcsk62
DOI: 10.1109/iccv.2017.371
515. Deep Inside Convolutional Networks: Visualising Image Classification Models and Saliency Maps
Karen Simonyan, Andrea Vedaldi, Andrew Zisserman
arXiv (2014-04-22) https://arxiv.org/abs/1312.6034
516. On Pixel-Wise Explanations for Non-Linear Classifier Decisions by Layer-Wise Relevance Propagation
Sebastian Bach, Alexander Binder, Grégoire Montavon, Frederick Klauschen, Klaus-Robert Müller, Wojciech Samek
PLOS ONE (2015-07-10) https://doi.org/gcgmcp
DOI: 10.1371/journal.pone.0130140 · PMID: 26161953 · PMCID: PMC4498753
517. Investigating the influence of noise and distractors on the interpretation of neural networks
Pieter-Jan Kindermans, Kristof Schütt, Klaus-Robert Müller, Sven Dähne
arXiv (2016-11-23) https://arxiv.org/abs/1611.07270
518. Striving for Simplicity: The All Convolutional Net
Jost Tobias Springenberg, Alexey Dosovitskiy, Thomas Brox, Martin Riedmiller
arXiv (2015-04-14) https://arxiv.org/abs/1412.6806
519. Salient Deconvolutional Networks
Aravindh Mahendran, Andrea Vedaldi
Lecture Notes in Computer Science (2016) https://doi.org/gcgk7p
DOI: 10.1007/978-3-319-46466-4_8
520. Grad-CAM: Visual Explanations from Deep Networks via Gradient-based Localization
Ramprasaath R. Selvaraju, Michael Cogswell, Abhishek Das, Ramakrishna Vedantam, Devi Parikh, Dhruv Batra
arXiv (2019-12-04) https://arxiv.org/abs/1610.02391
DOI: 10.1007/s11263-019-01228-7
521. Axiomatic Attribution for Deep Networks
Mukund Sundararajan, Ankur Taly, Qiqi Yan
arXiv (2017-06-14) https://arxiv.org/abs/1703.01365
522. An unexpected unity among methods for interpreting model predictions
Scott Lundberg, Su-In Lee
arXiv (2016-12-09) https://arxiv.org/abs/1611.07478
523. 17. A Value for n-Person Games
L. S. Shapley
Contributions to the Theory of Games (AM-28), Volume II (1953) https://doi.org/10.1515/9781400881970-018
DOI: 10.1515/9781400881970-018
524. Understanding Deep Image Representations by Inverting Them
Aravindh Mahendran, Andrea Vedaldi
arXiv (2014-12-02) https://arxiv.org/abs/1412.0035
525. Maximum entropy methods for extracting the learned features of deep neural networks
Alex Finnegan, Jun S. Song
bioRxiv (2017-10-05) https://doi.org/gcgk9w
DOI: 10.1101/105957
526. Visualizing Deep Convolutional Neural Networks Using Natural Pre-images
Aravindh Mahendran, Andrea Vedaldi
International Journal of Computer Vision (2016-05-18) https://doi.org/gcgk7x
DOI: 10.1007/s11263-016-0911-8
527. Inceptionism: Going Deeper into Neural Networks
Alexander Mordvintsev, Christopher Olah, Mike Tyka
Google Research Blog (2015-06) http://googleresearch.blogspot.co.uk/2015/06/inceptionism-going-deeper-into-neural.html
528. Visualizing Higher-Layer Features of a Deep Network
Dumitru Erhan, Yoshua Bengio, Aaron Courville, Pascal Vincent
University of Montreal (2009-06) http://www.iro.umontreal.ca/~lisa/publications2/index.php/publications/show/247
529. Understanding Neural Networks Through Deep Visualization
Jason Yosinski, Jeff Clune, Anh Nguyen, Thomas Fuchs, Hod Lipson
arXiv (2015-06-23) https://arxiv.org/abs/1506.06579
530. Neural Machine Translation by Jointly Learning to Align and Translate
Dzmitry Bahdanau, Kyunghyun Cho, Yoshua Bengio
arXiv (2016-05-23) https://arxiv.org/abs/1409.0473
531. Show, Attend and Tell: Neural Image Caption Generation with Visual Attention
Kelvin Xu, Jimmy Ba, Ryan Kiros, Kyunghyun Cho, Aaron Courville, Ruslan Salakhutdinov, Richard Zemel, Yoshua Bengio
arXiv (2016-04-20) https://arxiv.org/abs/1502.03044
532. Genetic Architect: Discovering Genomic Structure with Learned Neural Architectures
Laura Deming, Sasha Targ, Nate Sauder, Diogo Almeida, Chun Jimmie Ye
arXiv (2016-05-24) https://arxiv.org/abs/1605.07156
533. RETAIN: An Interpretable Predictive Model for Healthcare using Reverse Time Attention Mechanism
Edward Choi, Mohammad Taha Bahadori, Joshua A. Kulas, Andy Schuetz, Walter F. Stewart, Jimeng Sun
arXiv (2017-02-28) https://arxiv.org/abs/1608.05745
534. GRAM: Graph-based Attention Model for Healthcare Representation Learning
Edward Choi, Mohammad Taha Bahadori, Le Song, Walter F. Stewart, Jimeng Sun
arXiv (2017-04-04) https://arxiv.org/abs/1611.07012
535. Sequence learning with recurrent networks: analysis of internal representations
Joydeep Ghosh, Vijay Karamcheti
SPIE-Intl Soc Optical Eng (1992-07-01) https://doi.org/fbrtf4
DOI: 10.1117/12.140112
536. Visualizing and Understanding Recurrent Networks
Andrej Karpathy, Justin Johnson, Li Fei-Fei
arXiv (2015-11-18) https://arxiv.org/abs/1506.02078
537. LSTMVis: A Tool for Visual Analysis of Hidden State Dynamics in Recurrent Neural Networks
Hendrik Strobelt, Sebastian Gehrmann, Hanspeter Pfister, Alexander M. Rush
arXiv (2017-10-31) https://arxiv.org/abs/1606.07461
538. Automatic Rule Extraction from Long Short Term Memory Networks
W. James Murdoch, Arthur Szlam
arXiv (2017-02-28) https://arxiv.org/abs/1702.02540
539. Unsupervised Representation Learning with Deep Convolutional Generative
Adversarial Networks
Alec Radford, Luke Metz, Soumith Chintala
arXiv (2015-11-19) https://arxiv.org/abs/1511.06434v2
540. The Cancer Genome Atlas Pan-Cancer analysis project
John N Weinstein, Eric A Collisson, Gordon B Mills, Kenna R Mills Shaw, Brad A Ozenberger, Kyle Ellrott, Ilya Shmulevich, Chris Sander, Joshua M Stuart, The Cancer Genome Atlas Research Network
Nature Genetics (2013-09-26) https://doi.org/f3nt5c
DOI: 10.1038/ng.2764 · PMID: 24071849 · PMCID: PMC3919969
541. Extracting a Biologically Relevant Latent Space from Cancer Transcriptomes with Variational Autoencoders
Gregory P. Way, Casey S. Greene
bioRxiv (2017-10-02) https://doi.org/gcm44d
DOI: 10.1101/174474
542. Evaluating deep variational autoencoders trained on pan-cancer gene expression
Gregory P. Way, Casey S. Greene
arXiv (2017-11-15) https://arxiv.org/abs/1711.04828
543. GANs for Biological Image Synthesis
Anton Osokin, Anatole Chessel, Rafael E. Carazo Salas, Federico Vaggi
arXiv (2017-09-13) https://arxiv.org/abs/1708.04692
544. CytoGAN: Generative Modeling of Cell Images
Peter Goldsborough, Nick Pawlowski, Juan C Caicedo, Shantanu Singh, Anne E Carpenter
bioRxiv (2017-12-02) https://doi.org/gcm44f
DOI: 10.1101/227645
545. Understanding Black-box Predictions via Influence Functions
Pang Wei Koh, Percy Liang
arXiv (2017-07-11) https://arxiv.org/abs/1703.04730
546. ActiVis: Visual Exploration of Industry-Scale Deep Neural Network Models
Minsuk Kahng, Pierre Y. Andrews, Aditya Kalro, Duen Horng Chau
arXiv (2017-08-10) https://arxiv.org/abs/1704.01942
547. Towards Better Analysis of Deep Convolutional Neural Networks
Mengchen Liu, Jiaxin Shi, Zhen Li, Chongxuan Li, Jun Zhu, Shixia Liu
arXiv (2016-05-05) https://arxiv.org/abs/1604.07043
548. Distilling Knowledge from Deep Networks with Applications to Healthcare Domain
Zhengping Che, Sanjay Purushotham, Robinder Khemani, Yan Liu
arXiv (2015-12-14) https://arxiv.org/abs/1512.03542
549. Rationalizing Neural Predictions
Tao Lei, Regina Barzilay, Tommi Jaakkola
arXiv (2016-11-04) https://arxiv.org/abs/1606.04155
550. Learning multiple layers of features from tiny images
Alex Krizhevsky
(2009) https://www.cs.toronto.edu/~kriz/learning-features-2009-TR.pdf
551. Functional Knowledge Transfer for High-accuracy Prediction of Under-studied Biological Processes
Christopher Y. Park, Aaron K. Wong, Casey S. Greene, Jessica Rowland, Yuanfang Guan, Lars A. Bongo, Rebecca D. Burdine, Olga G. Troyanskaya
PLoS Computational Biology (2013-03-14) https://doi.org/f4qtp9
DOI: 10.1371/journal.pcbi.1002957 · PMID: 23516347 · PMCID: PMC3597527
552. DeepAD: Alzheimer’s Disease Classification via Deep Convolutional Neural Networks using MRI and fMRI
Saman Sarraf, Danielle D. DeSouza, John Anderson, Ghassem Tofighi, for the Alzheimer’s Disease Neuroimaging Initiativ
bioRxiv (2017-01-14) https://doi.org/gcgk9b
DOI: 10.1101/070441
553. DeepBound: Accurate Identification of Transcript Boundaries via Deep Convolutional Neural Fields
Mingfu Shao, Jianzhu Ma, Sheng Wang
bioRxiv (2017-04-07) https://doi.org/gcgk92
DOI: 10.1101/125229
554. A general framework for estimating the relative pathogenicity of human genetic variants
Martin Kircher, Daniela M Witten, Preti Jain, Brian J O’Roak, Gregory M Cooper, Jay Shendure
Nature Genetics (2014-02-02) https://doi.org/f5s57j
DOI: 10.1038/ng.2892 · PMID: 24487276 · PMCID: PMC3992975
555. Diet Networks: Thin Parameters for Fat Genomics
Adriana Romero, Pierre Luc Carrier, Akram Erraqabi, Tristan Sylvain, Alex Auvolat, Etienne Dejoie, Marc-André Legault, Marie-Pierre Dubé, Julie G. Hussin, Yoshua Bengio
International Conference on Learning Representations 2017 (2016-11-04) https://openreview.net/forum?id=Sk-oDY9ge¬eId=Sk-oDY9ge
556. Deep learning in neural networks: An overview
Jürgen Schmidhuber
Neural Networks (2015-01) https://doi.org/f6v78n
DOI: 10.1016/j.neunet.2014.09.003 · PMID: 25462637
557. Deep Learning with Limited Numerical Precision
Suyog Gupta, Ankur Agrawal, Kailash Gopalakrishnan, Pritish Narayanan
arXiv (2015-02-11) https://arxiv.org/abs/1502.02551
558. Training deep neural networks with low precision multiplications
Matthieu Courbariaux, Yoshua Bengio, Jean-Pierre David
arXiv (2015-09-24) https://arxiv.org/abs/1412.7024
559. Taming the Wild: A Unified Analysis of Hogwild!-Style Algorithms
Christopher De Sa, Ce Zhang, Kunle Olukotun, Christopher Ré
Advances in neural information processing systems (2015-12) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4907892/
PMID: 27330264 · PMCID: PMC4907892
560. Quantized Neural Networks: Training Neural Networks with Low Precision Weights and Activations
Itay Hubara, Matthieu Courbariaux, Daniel Soudry, Ran El-Yaniv, Yoshua Bengio
arXiv (2016-09-23) https://arxiv.org/abs/1609.07061
561. Do Deep Nets Really Need to be Deep?
Lei Jimmy Ba, Rich Caruana
arXiv (2014-10-14) https://arxiv.org/abs/1312.6184
562. Distilling the Knowledge in a Neural Network
Geoffrey Hinton, Oriol Vinyals, Jeff Dean
arXiv (2015-03-10) https://arxiv.org/abs/1503.02531
563. Large-scale deep unsupervised learning using graphics processors
Rajat Raina, Anand Madhavan, Andrew Y. Ng
Association for Computing Machinery (ACM) (2009) https://doi.org/dfh65x
DOI: 10.1145/1553374.1553486
564. Improving the speed of neural networks on CPUs
Vincent Vanhoucke, Andrew Senior, Mark Z. Mao
Google Research (2011) https://research.google/pubs/pub37631/
565. On parallelizability of stochastic gradient descent for speech DNNS
Frank Seide, Hao Fu, Jasha Droppo, Gang Li, Dong Yu
Institute of Electrical and Electronics Engineers (IEEE) (2014-05) https://doi.org/gcgk99
DOI: 10.1109/icassp.2014.6853593
566. Caffe con Troll: Shallow Ideas to Speed Up Deep Learning
Stefan Hadjis, Firas Abuzaid, Ce Zhang, Christopher Ré
arXiv (2015-05-28) https://arxiv.org/abs/1504.04343
567. Growing pains for deep learning
Chris Edwards
Communications of the ACM (2015-06-25) https://doi.org/gcgmbz
DOI: 10.1145/2771283
568. Experiments on Parallel Training of Deep Neural Network using Model Averaging
Hang Su, Haoyu Chen
arXiv (2018-07-03) https://arxiv.org/abs/1507.01239
569. Efficient mini-batch training for stochastic optimization
Mu Li, Tong Zhang, Yuqiang Chen, Alexander J. Smola
Association for Computing Machinery (ACM) (2014) https://doi.org/gcgmbw
DOI: 10.1145/2623330.2623612
570. CGBVS‐DNN: Prediction of Compound‐protein Interactions Based on Deep Learning
Masatoshi Hamanaka, Kei Taneishi, Hiroaki Iwata, Jun Ye, Jianguo Pei, Jinlong Hou, Yasushi Okuno
Molecular Informatics (2016-08-12) https://doi.org/f3q59v
DOI: 10.1002/minf.201600045 · PMID: 27515489
571. cuDNN: Efficient Primitives for Deep Learning
Sharan Chetlur, Cliff Woolley, Philippe Vandermersch, Jonathan Cohen, John Tran, Bryan Catanzaro, Evan Shelhamer
arXiv (2014-12-19) https://arxiv.org/abs/1410.0759
572. Compressing Neural Networks with the Hashing Trick
Wenlin Chen, James T. Wilson, Stephen Tyree, Kilian Q. Weinberger, Yixin Chen
arXiv (2015-04-21) https://arxiv.org/abs/1504.04788
573. Deep Learning on FPGAs: Past, Present, and Future
Griffin Lacey, Graham W. Taylor, Shawki Areibi
arXiv (2016-02-16) https://arxiv.org/abs/1602.04283
574. In-Datacenter Performance Analysis of a Tensor Processing Unit
Norman P. Jouppi, Cliff Young, Nishant Patil, David Patterson, Gaurav Agrawal, Raminder Bajwa, Sarah Bates, Suresh Bhatia, Nan Boden, Al Borchers, … Doe Hyun Yoon
arXiv (2017-04-18) https://arxiv.org/abs/1704.04760
575. MapReduce
Jeffrey Dean, Sanjay Ghemawat
Communications of the ACM (2008-01-01) https://doi.org/br3wxw
DOI: 10.1145/1327452.1327492
576. Distributed GraphLab
Yucheng Low, Danny Bickson, Joseph Gonzalez, Carlos Guestrin, Aapo Kyrola, Joseph M. Hellerstein
Proceedings of the VLDB Endowment (2012-04-01) https://doi.org/gcgmcs
DOI: 10.14778/2212351.2212354
577. Large Scale Distributed Deep Networks
Jeffrey Dean, Greg S Corrado, Rajat Monga, Kai Chen, Matthieu Devin, Quoc V Le, Mark Z Mao, Marc’Aurelio Ranzato, Andrew Senior, Paul Tucker, … Andrew Y Ng
Neural Information Processing Systems 2012 (2012-12) http://research.google.com/archive/large_deep_networks_nips2012.html
578. SparkNet: Training Deep Networks in Spark
Philipp Moritz, Robert Nishihara, Ion Stoica, Michael I. Jordan
arXiv (2016-03-01) https://arxiv.org/abs/1511.06051
579. MLlib: Machine Learning in Apache Spark
Xiangrui Meng, Joseph Bradley, Burak Yavuz, Evan Sparks, Shivaram Venkataraman, Davies Liu, Jeremy Freeman, DB Tsai, Manish Amde, Sean Owen, … Ameet Talwalkar
arXiv (2015-05-27) https://arxiv.org/abs/1505.06807
580. TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems
Martín Abadi, Ashish Agarwal, Paul Barham, Eugene Brevdo, Zhifeng Chen, Craig Citro, Greg S. Corrado, Andy Davis, Jeffrey Dean, Matthieu Devin, … Xiaoqiang Zheng
arXiv (2016-03-17) https://arxiv.org/abs/1603.04467
581. fchollet/keras
GitHub
(2017) https://github.com/fchollet/keras
582. maxpumperla/elephas
GitHub
(2017) https://github.com/maxpumperla/elephas
583. Deep learning with COTS HPC systems
Adam Coates, Brody Huval, Tao Wang, David Wu, Bryan Catanzaro, Ng Andrew
International Conference on Machine Learning (2013-02-13) http://proceedings.mlr.press/v28/coates13.html
584. Ensemble-Compression: A New Method for Parallel Training of Deep Neural Networks
Shizhao Sun, Wei Chen, Jiang Bian, Xiaoguang Liu, Tie-Yan Liu
arXiv (2017-07-19) https://arxiv.org/abs/1606.00575
585. Algorithms for Hyper-parameter Optimization
James Bergstra, Rémi Bardenet, Yoshua Bengio, Balázs Kégl
Proceedings of the 24th International Conference on Neural Information Processing Systems (2011) http://dl.acm.org/citation.cfm?id=2986459.2986743
ISBN: 978-1-61839-599-3
586. Random Search for Hyper-Parameter Optimization
James Bergstra, Yoshua Bengio
Journal of Machine Learning Research (2012) http://www.jmlr.org/papers/v13/bergstra12a.html
587. Cloud computing and the DNA data race
Michael C Schatz, Ben Langmead, Steven L Salzberg
Nature Biotechnology (2010-07) https://doi.org/cskgd3
DOI: 10.1038/nbt0710-691 · PMID: 20622843 · PMCID: PMC2904649
588. The real cost of sequencing: scaling computation to keep pace with data generation
Paul Muir, Shantao Li, Shaoke Lou, Daifeng Wang, Daniel J Spakowicz, Leonidas Salichos, Jing Zhang, George M. Weinstock, Farren Isaacs, Joel Rozowsky, Mark Gerstein
Genome Biology (2016-03-23) https://doi.org/gcgmcb
DOI: 10.1186/s13059-016-0917-0 · PMID: 27009100 · PMCID: PMC4806511
589. The case for cloud computing in genome informatics
Lincoln D Stein
Genome Biology (2010) https://doi.org/ach
DOI: 10.1186/gb-2010-11-5-207 · PMID: 20441614 · PMCID: PMC2898083
590. One weird trick for parallelizing convolutional neural networks
Alex Krizhevsky
arXiv (2014-04-29) https://arxiv.org/abs/1404.5997
591. A view of cloud computing
Michael Armbrust, Ion Stoica, Matei Zaharia, Armando Fox, Rean Griffith, Anthony D. Joseph, Randy Katz, Andy Konwinski, Gunho Lee, David Patterson, Ariel Rabkin
Communications of the ACM (2010-04-01) https://doi.org/c4svd8
DOI: 10.1145/1721654.1721672
592. Data Sharing
Dan L. Longo, Jeffrey M. Drazen
New England Journal of Medicine (2016-01-21) https://doi.org/gcgk8r
DOI: 10.1056/nejme1516564 · PMID: 26789876
593. Celebrating parasites
Casey S Greene, Lana X Garmire, Jack A Gilbert, Marylyn D Ritchie, Lawrence E Hunter
Nature Genetics (2017-04-01) https://doi.org/gcgk8d
DOI: 10.1038/ng.3830 · PMID: 28358134 · PMCID: PMC5710834
594. Is Multitask Deep Learning Practical for Pharma?
Bharath Ramsundar, Bowen Liu, Zhenqin Wu, Andreas Verras, Matthew Tudor, Robert P. Sheridan, Vijay Pande
Journal of Chemical Information and Modeling (2017-08) https://doi.org/gcnzrm
DOI: 10.1021/acs.jcim.7b00146 · PMID: 28692267
595. Enhancing reproducibility for computational methods
V. Stodden, M. McNutt, D. H. Bailey, E. Deelman, Y. Gil, B. Hanson, M. A. Heroux, J. P. A. Ioannidis, M. Taufer
Science (2016-12-08) https://doi.org/gbr42b
DOI: 10.1126/science.aah6168 · PMID: 27940837
596. DragoNN http://kundajelab.github.io/dragonn/
597. How transferable are features in deep neural networks?
Jason Yosinski, Jeff Clune, Yoshua Bengio, Hod Lipson
Advances in Neural Information Processing Systems 27 (2014) http://papers.nips.cc/paper/5347-how-transferable-are-features-in-deep-neural-networks.pdf
598. Deep Model Based Transfer and Multi-Task Learning for Biological Image Analysis
Wenlu Zhang, Rongjian Li, Tao Zeng, Qian Sun, Sudhir Kumar, Jieping Ye, Shuiwang Ji
Association for Computing Machinery (ACM) (2015) https://doi.org/gcgmb2
DOI: 10.1145/2783258.2783304
599. Deep convolutional neural networks for annotating gene expression patterns in the mouse brain
Tao Zeng, Rongjian Li, Ravi Mukkamala, Jieping Ye, Shuiwang Ji
BMC Bioinformatics (2015-05-07) https://doi.org/gb8w84
DOI: 10.1186/s12859-015-0553-9 · PMID: 25948335 · PMCID: PMC4432953
600. Accurate Classification of Protein Subcellular Localization from High-Throughput Microscopy Images Using Deep Learning
Tanel Pärnamaa, Leopold Parts
G3: Genes|Genomes|Genetics (2017-04-08) https://doi.org/10.1534/g3.116.033654
DOI: 10.1534/g3.116.033654
601. Automated analysis of high‐content microscopy data with deep learning
Oren Z Kraus, Ben T Grys, Jimmy Ba, Yolanda Chong, Brendan J Frey, Charles Boone, Brenda J Andrews
Molecular Systems Biology (2017-04-18) https://doi.org/f93cpr
DOI: 10.15252/msb.20177551 · PMID: 28420678 · PMCID: PMC5408780
602. Multimodal Deep Learning
Jiquan Ngiam, Aditya Khosla, Mingyu Kim, Juhan Nam, Honglak Lee, Andrew Y. Ng
Proceedings of the 28th International Conference on Machine Learning (2011) https://ccrma.stanford.edu/~juhan/pubs/NgiamKhoslaKimNamLeeNg2011.pdf
603. Deep Learning based multi-omics integration robustly predicts survival in liver cancer
Kumardeep Chaudhary, Olivier B. Poirion, Liangqun Lu, Lana X. Garmire
bioRxiv (2017-09-18) https://doi.org/gcgk9z
DOI: 10.1101/114892
604. FIDDLE: An integrative deep learning framework for functional genomic data inference
Umet Eser, L. Stirling Churchman
bioRxiv (2016-10-17) https://doi.org/gcgk9g
DOI: 10.1101/081380
605. Modeling Reactivity to Biological Macromolecules with a Deep Multitask Network
Tyler B. Hughes, Na Le Dang, Grover P. Miller, S. Joshua Swamidass
ACS Central Science (2016-07-29) https://doi.org/gcgk78
DOI: 10.1021/acscentsci.6b00162 · PMID: 27610414 · PMCID: PMC4999971
606. IBM edges closer to human speech recognition
BI Intelligence
Business Insider (2017-03-14) http://www.businessinsider.com/ibm-edges-closer-to-human-speech-recognition-2017-3
607. Achieving Human Parity in Conversational Speech Recognition
W. Xiong, J. Droppo, X. Huang, F. Seide, M. Seltzer, A. Stolcke, D. Yu, G. Zweig
arXiv (2018-12-06) https://arxiv.org/abs/1610.05256
608. English Conversational Telephone Speech Recognition by Humans and Machines
George Saon, Gakuto Kurata, Tom Sercu, Kartik Audhkhasi, Samuel Thomas, Dimitrios Dimitriadis, Xiaodong Cui, Bhuvana Ramabhadran, Michael Picheny, Lynn-Li Lim, … Phil Hall
arXiv (2017-03-08) https://arxiv.org/abs/1703.02136
609. Intriguing properties of neural networks
Christian Szegedy, Wojciech Zaremba, Ilya Sutskever, Joan Bruna, Dumitru Erhan, Ian Goodfellow, Rob Fergus
arXiv (2014-02-20) https://arxiv.org/abs/1312.6199
610. Explaining and Harnessing Adversarial Examples
Ian J. Goodfellow, Jonathon Shlens, Christian Szegedy
arXiv (2015-03-24) https://arxiv.org/abs/1412.6572
611. Towards the Science of Security and Privacy in Machine Learning
Nicolas Papernot, Patrick McDaniel, Arunesh Sinha, Michael Wellman
arXiv (2016-11-14) https://arxiv.org/abs/1611.03814
612. Feature Squeezing: Detecting Adversarial Examples in Deep Neural Networks
Weilin Xu, David Evans, Yanjun Qi
arXiv (2017-12-07) https://arxiv.org/abs/1704.01155
DOI: 10.14722/ndss.2018.23198
613. Open collaborative writing with Manubot
Daniel S. Himmelstein, Vincent Rubinetti, David R. Slochower, Dongbo Hu, Venkat S. Malladi, Casey S. Greene, Anthony Gitter
PLOS Computational Biology (2019-06-24) https://doi.org/c7np
DOI: 10.1371/journal.pcbi.1007128 · PMID: 31233491 · PMCID: PMC6611653
615. greenelab/deep-review
GitHub
(2017) https://github.com/greenelab/deep-review