Summary
BioBombe examines many low-dimensional representations of gene expression data. Named after the large mechanical devices built by Alan Turing and other cryptologists during World War II to decode secret messages, BioBombe represents an approach to decipher hidden messages embedded in gene expression data. In the manuscript, we use this approach to compare the biological representations learned by various compression algorithms across different latent space dimensionalities ranging from k = 2 to k = 200.
This website provides convenient links to all the resources produced for the manuscript including software, processed gene expression input data, compressed features for all algorithms, latent dimensionalities, and initializations for all datasets, and performance results and models for the large scale cancer-type and mutation classification analysis.
Primary Findings
There does not exist a single optimal algorithm or latent dimensionality for learning biological representations. Many different biological signatures are learned by different compression algorithms at various latent dimensionalities. A practitioner aiming to optimize feature discovery by compressing gene expression data should use multiple algorithms across a large range of latent dimensionalities.
Citation
Sequential compression of gene expression across dimensionalities
and methods reveals no single best method or dimensionality
Way, G.P., Zietz, M., Rubinetti, V., Himmelstein, D.S., Greene, C.S.
biorXiv preprint (2019) ·
doi:10.1101/573782
Approach
We train five compression algorithms (principal components analysis (PCA), independent components analysis (ICA), non-negative matrix factorization (NMF), denoising autoencoders (DAE), and variational autoencoders (VAE)) using three benchmark gene expression datasets: The Cancer Genome Atlas (TCGA), Genome Tissue Expression Consortium project (GTEx), and the Therapeutically Applicable Research To Generate Effective Treatments (TARGET) project across a wide range of latent dimensionalities (k). We assess model performance by measuring reconstruction, correlation between input and reconstructed output, model stability and geneset coverage.
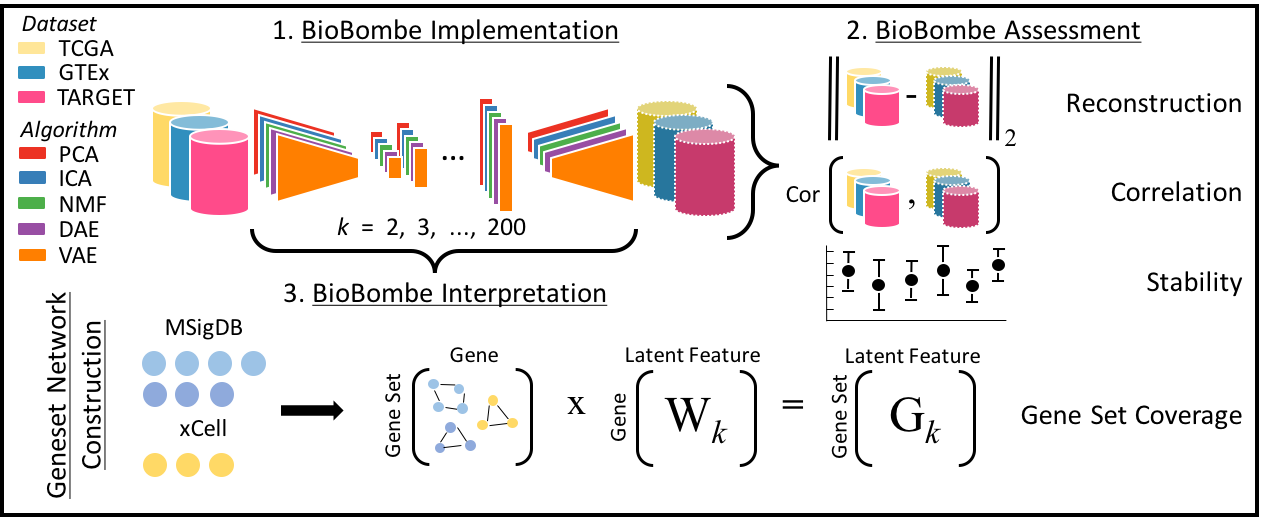
Resources
Software
Includes code, data, documentation, results, figures, and a computational environment for the full analysis. Each numbered module represents specific data processing steps or analysis results.
Input Data
Includes Processed Training and Testing Datasets for TCGA, GTEx, and Target as git LFS files
Heterogeneous Networks
Includes real and permuted hetnets1 for MSigDB2 and xCell3 gene sets, as well as more details about heterogeneous networks.
1
Systematic integration of biomedical knowledge prioritizes drugs
for repurposing
Himmelstein DS, Lizee A, Hessler C, Brueggeman L, Chen SL, Hadley D,
Green A, Khankhanian P, Baranzini SE.
eLife (2017) ·
doi:10.7554/eLife.26726
2
The Molecular Signatures Database Hallmark Gene Set Collection
Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov
JP, Tamayo P.
Cell Systems (2015) · 1:417-25
3
xCell: digitally portraying the tissue cellular heterogeneity
landscape
Aran D, Hu Z, Butte AJ.
Genome Biology (2017) ·
doi:10.1186/s13059-017-1349-1
Compressed Features
Results:
Randomly Permuted Data:
TCGA Classification Results
Includes BioBombe feature coefficients, sample activation scores, and classifier metrics for all supervised learning models (elastic net logistic regression) trained to predict cancer-type and mutation status.
Acknowledge
This work was funded in party by The Gordon and Betty Moore Foundation under GBMF 4552 (CSG) and the National Institutes of Health's National Human Genome Research Institute under R01 HG010067 (CSG) and the National Institutes of Health under T32 HG000046 (GPW).
We would like to thank Jaclyn Taroni, Yoson Park, and Alexandra Lee for insightful discussions and code review. We also thank Jo Lynne Rokita and John Maris for insightful discussions regarding the neuroblastoma analysis.